

Inference of Coalescence Times and Variant Ages Using Convolutional Neural Networks
- PMID: 37738175
- PMCID: PMC10581698
- DOI: 10.1093/molbev/msad211
Inference of Coalescence Times and Variant Ages Using Convolutional Neural Networks
Abstract
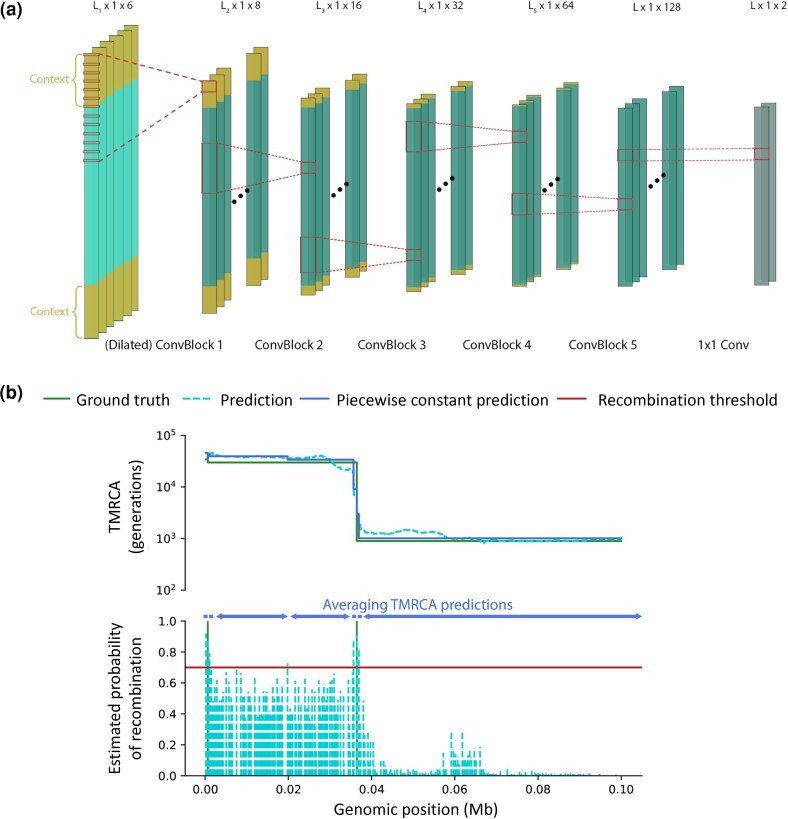
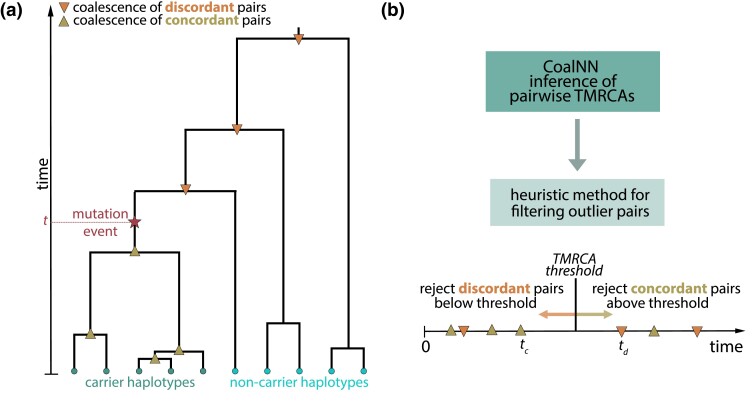
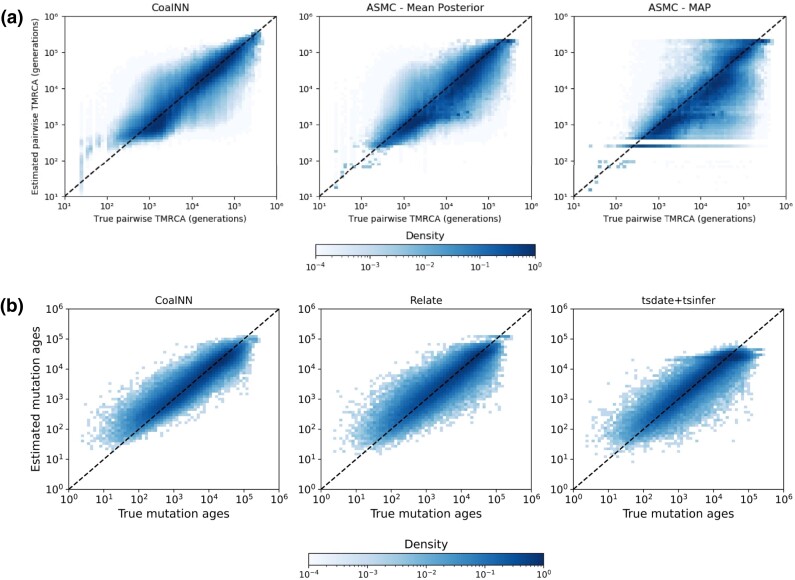
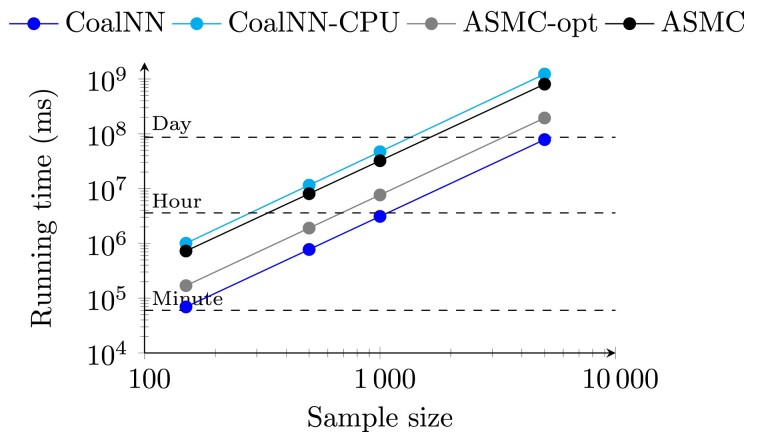
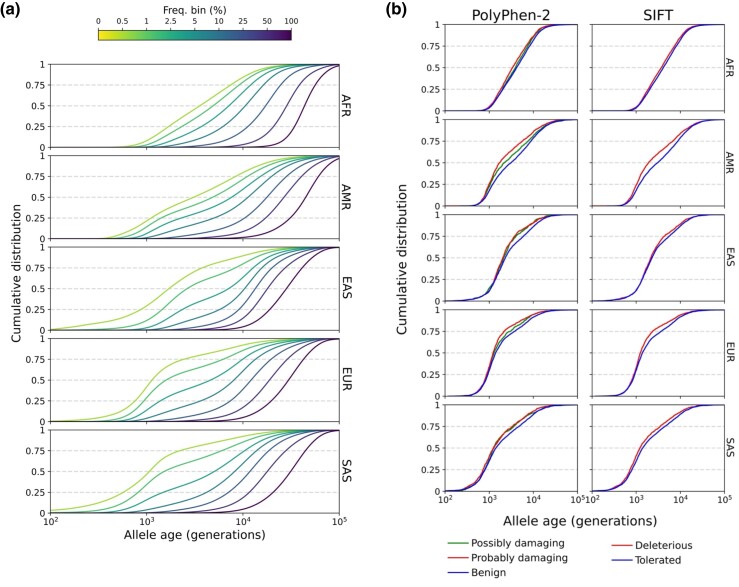
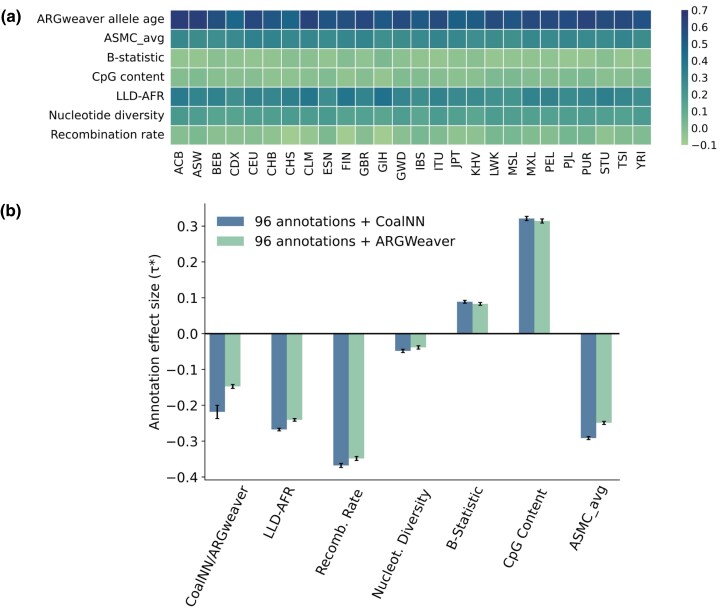
Accurate inference of the time to the most recent common ancestor (TMRCA) between pairs of individuals and of the age of genomic variants is key in several population genetic analyses. We developed a likelihood-free approach, called CoalNN, which uses a convolutional neural network to predict pairwise TMRCAs and allele ages from sequencing or SNP array data. CoalNN is trained through simulation and can be adapted to varying parameters, such as demographic history, using transfer learning. Across several simulated scenarios, CoalNN matched or outperformed the accuracy of model-based approaches for pairwise TMRCA and allele age prediction. We applied CoalNN to settings for which model-based approaches are under-developed and performed analyses to gain insights into the set of features it uses to perform TMRCA prediction. We next used CoalNN to analyze 2,504 samples from 26 populations in the 1,000 Genome Project data set, inferring the age of ∼80 million variants. We observed substantial variation across populations and for variants predicted to be pathogenic, reflecting heterogeneous demographic histories and the action of negative selection. We used CoalNN's predicted allele ages to construct genome-wide annotations capturing the signature of past negative selection. We performed LD-score regression analysis of heritability using summary association statistics from 63 independent complex traits and diseases (average N=314k), observing increased annotation-specific effects on heritability compared to a previous allele age annotation. These results highlight the effectiveness of using likelihood-free, simulation-trained models to infer properties of gene genealogies in large genomic data sets.
Keywords: allele age; coalescence time; heritability; machine learning; natural selection.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
