

EGR1-mediated metabolic reprogramming to oxidative phosphorylation contributes to ibrutinib resistance in B-cell lymphoma
- PMID: 37738652
- PMCID: PMC10731920
- DOI: 10.1182/blood.2023020142
EGR1-mediated metabolic reprogramming to oxidative phosphorylation contributes to ibrutinib resistance in B-cell lymphoma
Abstract
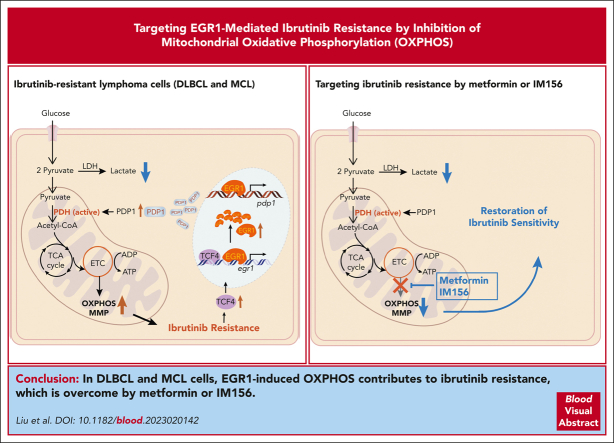
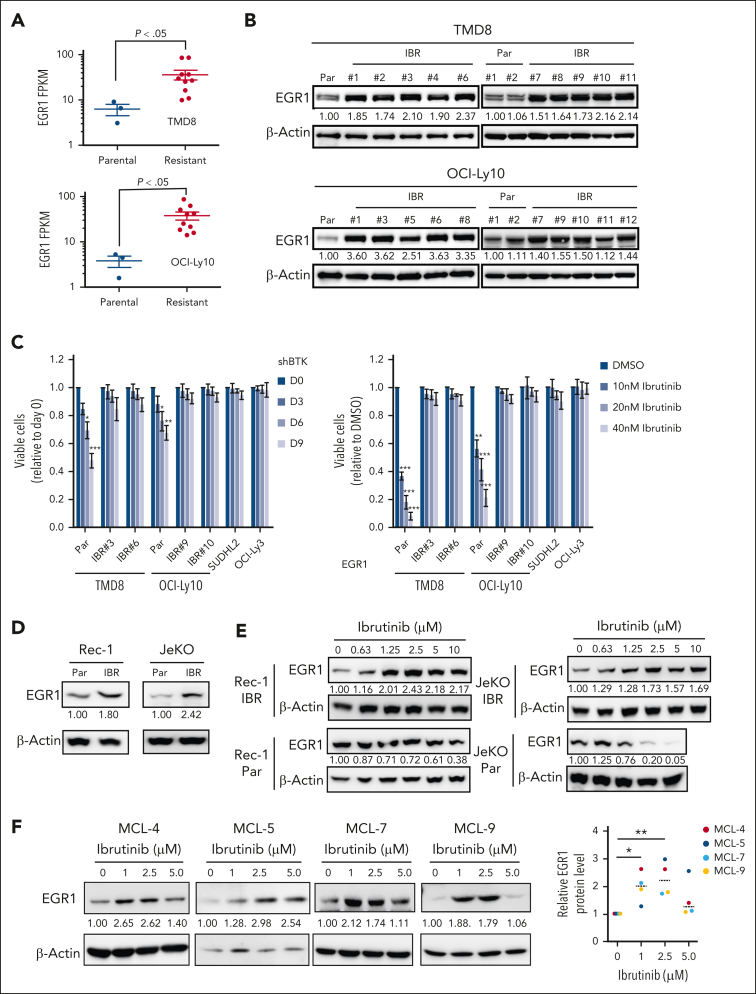
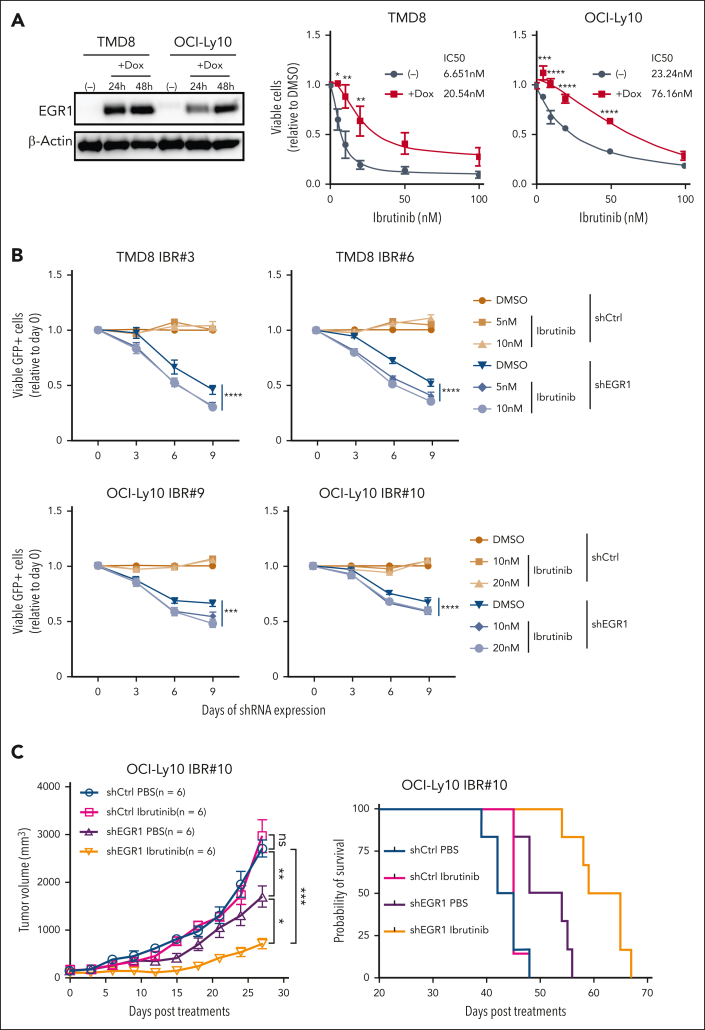
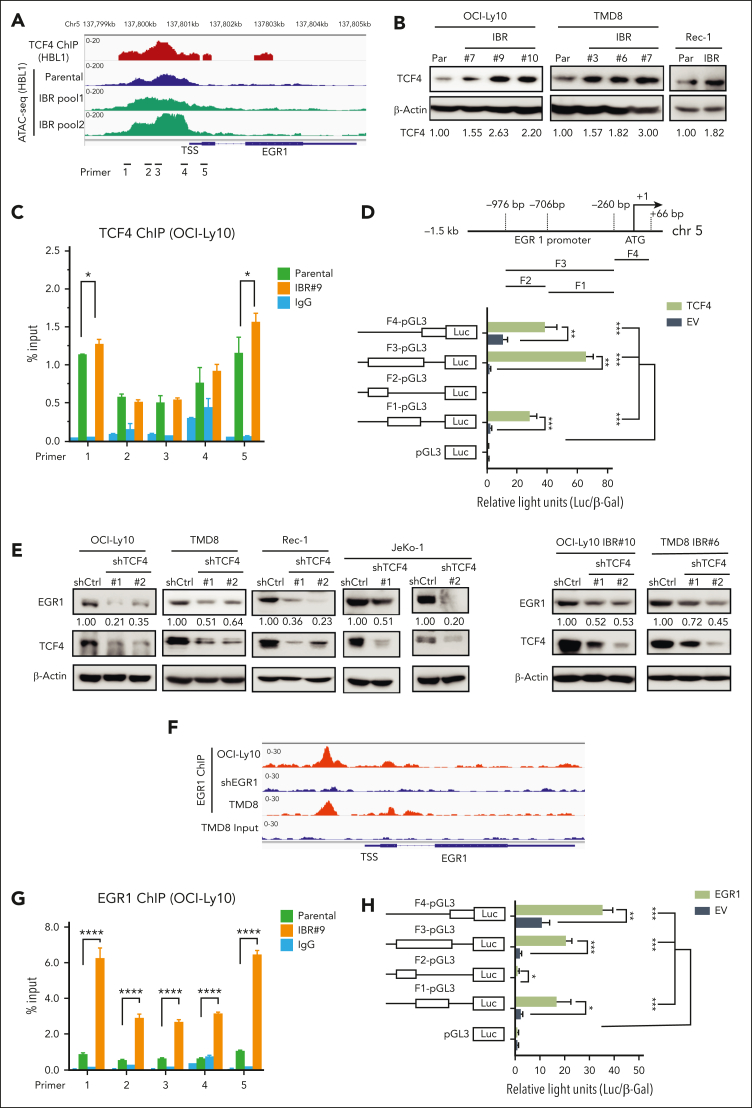
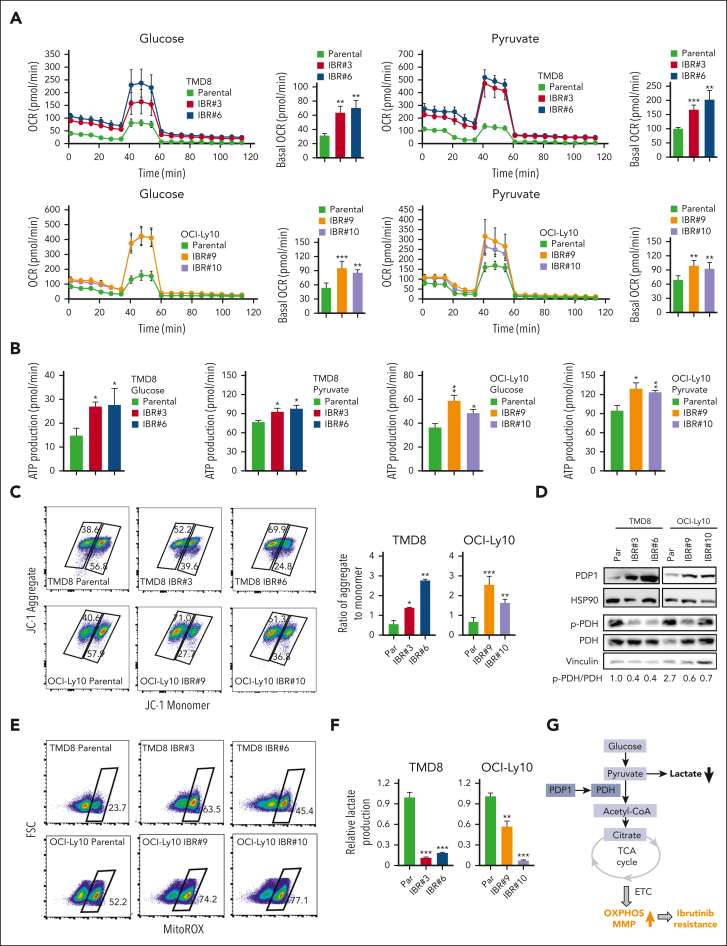
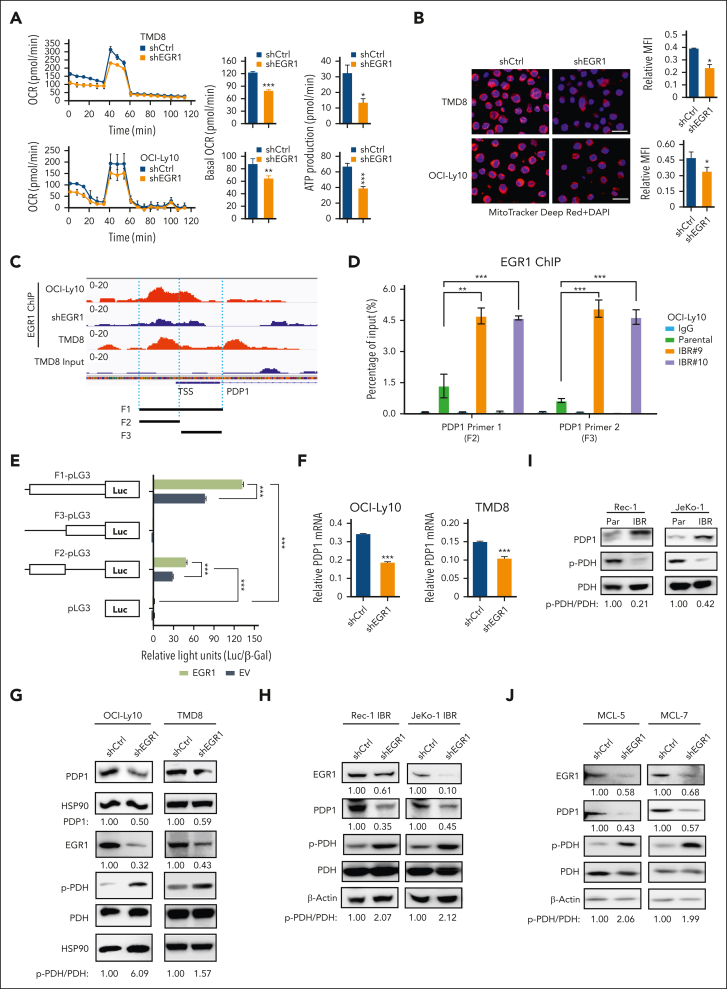
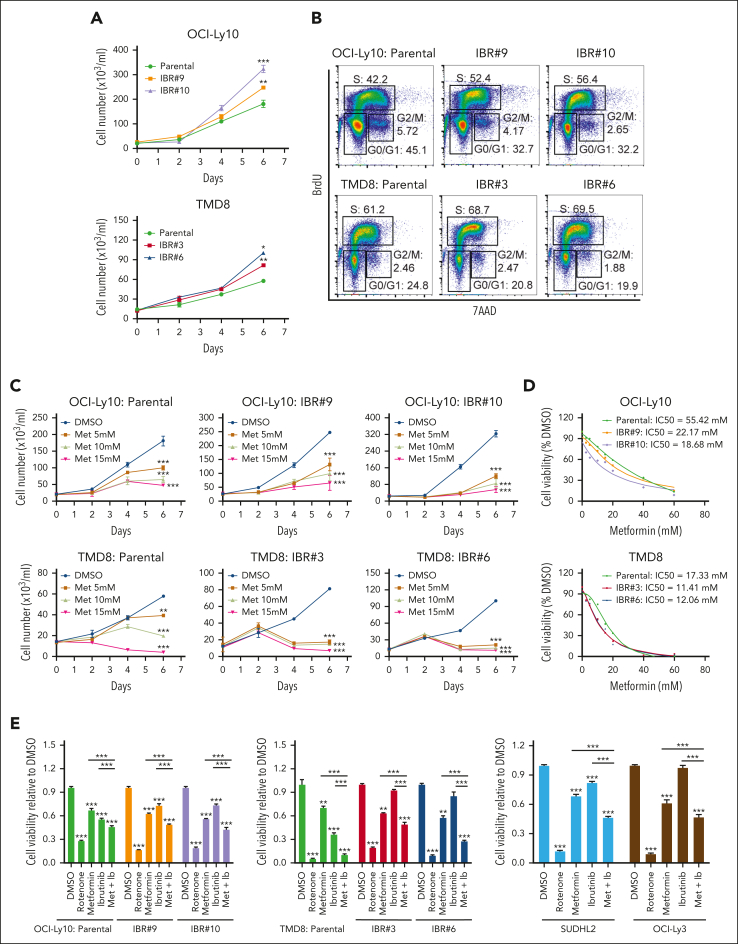
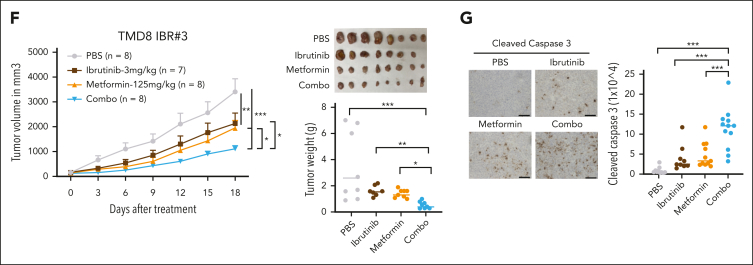
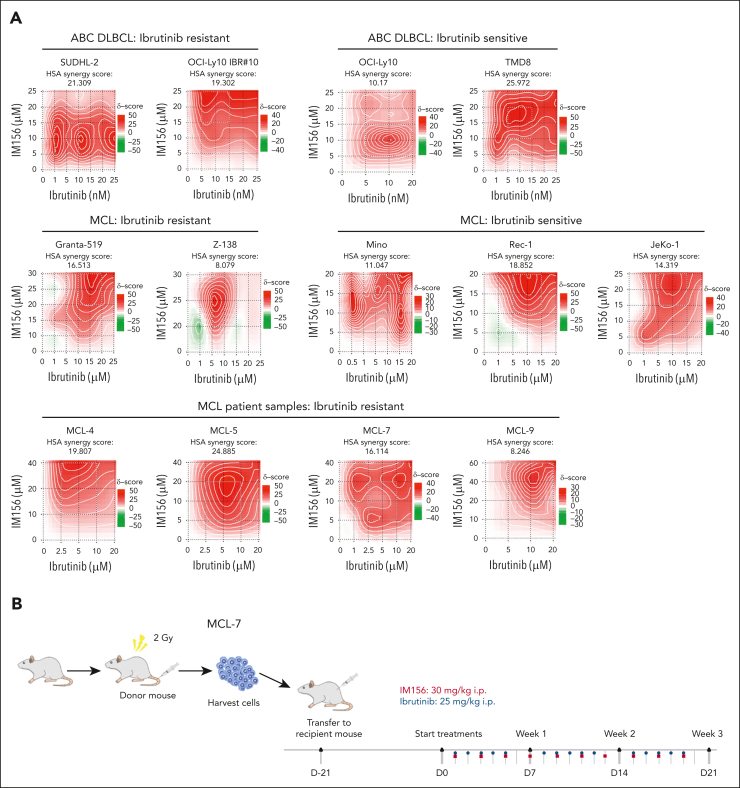
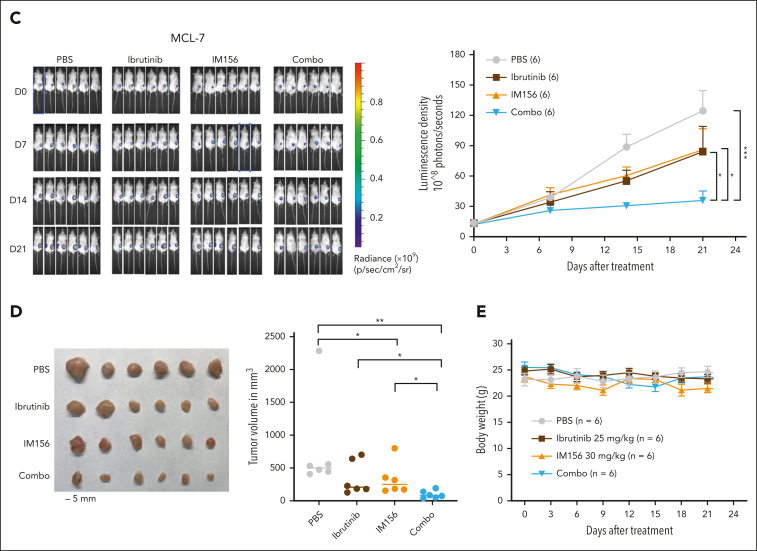
The use of Bruton tyrosine kinase inhibitors, such as ibrutinib, to block B-cell receptor signaling has achieved a remarkable clinical response in several B-cell malignancies, including mantle cell lymphoma (MCL) and diffuse large B-cell lymphoma (DLBCL). Acquired drug resistance, however, is significant and affects the long-term survival of these patients. Here, we demonstrate that the transcription factor early growth response gene 1 (EGR1) is involved in ibrutinib resistance. We found that EGR1 expression is elevated in ibrutinib-resistant activated B-cell-like subtype DLBCL and MCL cells and can be further upregulated upon ibrutinib treatment. Genetic and pharmacological analyses revealed that overexpressed EGR1 mediates ibrutinib resistance. Mechanistically, TCF4 and EGR1 self-regulation induce EGR1 overexpression that mediates metabolic reprogramming to oxidative phosphorylation (OXPHOS) through the transcriptional activation of PDP1, a phosphatase that dephosphorylates and activates the E1 component of the large pyruvate dehydrogenase complex. Therefore, EGR1-mediated PDP1 activation increases intracellular adenosine triphosphate production, leading to sufficient energy to enhance the proliferation and survival of ibrutinib-resistant lymphoma cells. Finally, we demonstrate that targeting OXPHOS with metformin or IM156, a newly developed OXPHOS inhibitor, inhibits the growth of ibrutinib-resistant lymphoma cells both in vitro and in a patient-derived xenograft mouse model. These findings suggest that targeting EGR1-mediated metabolic reprogramming to OXPHOS with metformin or IM156 provides a potential therapeutic strategy to overcome ibrutinib resistance in relapsed/refractory DLBCL or MCL.
Conflict of interest statement
Conflict-of-interest disclosure: C.M.C. receives honorarium for advisory board membership for Bayer, Elephas, Nektar Therapeutics, Novartis, and WiCell Research Institute, which had no input in the study design, analysis, manuscript preparation, or decision to submit for publication. The remaining authors declare no competing financial interests.
Thomas A. Waldmann died on 25 September 2021.
Figures
Comment in
-
EGR1 changes course in B-cell lymphoma.Blood. 2023 Nov 30;142(22):1850-1851. doi: 10.1182/blood.2023022493. Blood. 2023. PMID: 38032671 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
