

Epigenetic reprogramming of a distal developmental enhancer cluster drives SOX2 overexpression in breast and lung adenocarcinoma
- PMID: 37738673
- PMCID: PMC10602899
- DOI: 10.1093/nar/gkad734
Epigenetic reprogramming of a distal developmental enhancer cluster drives SOX2 overexpression in breast and lung adenocarcinoma
Abstract
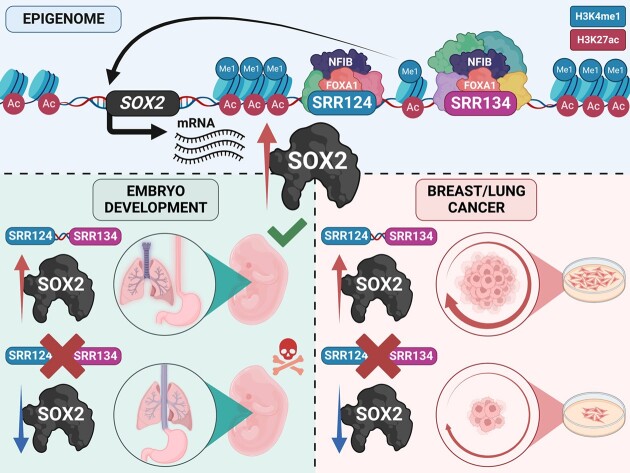
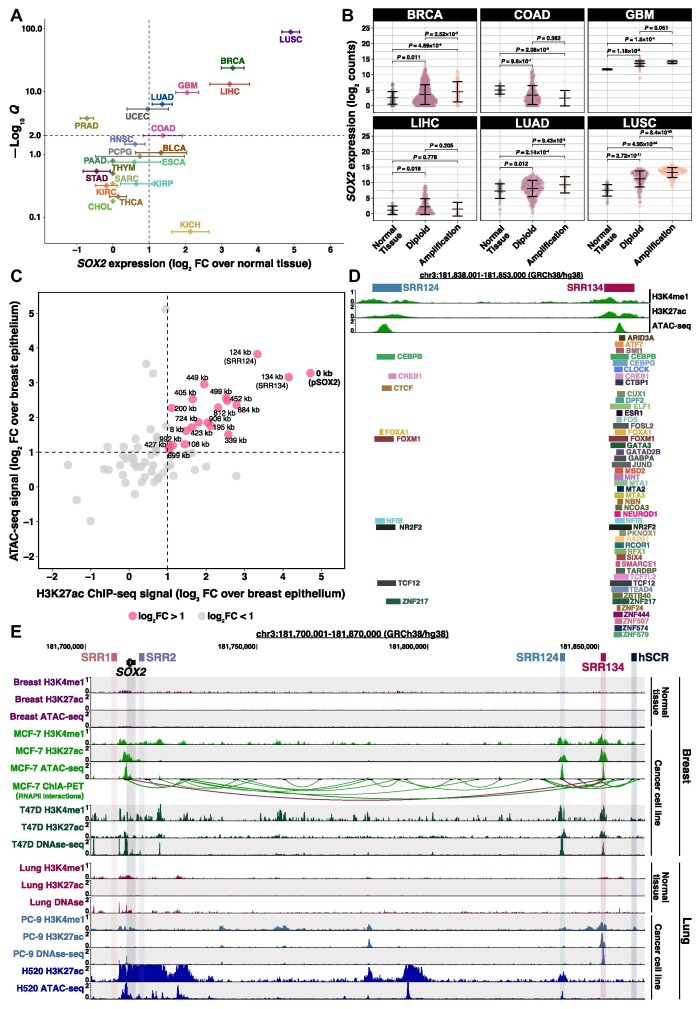
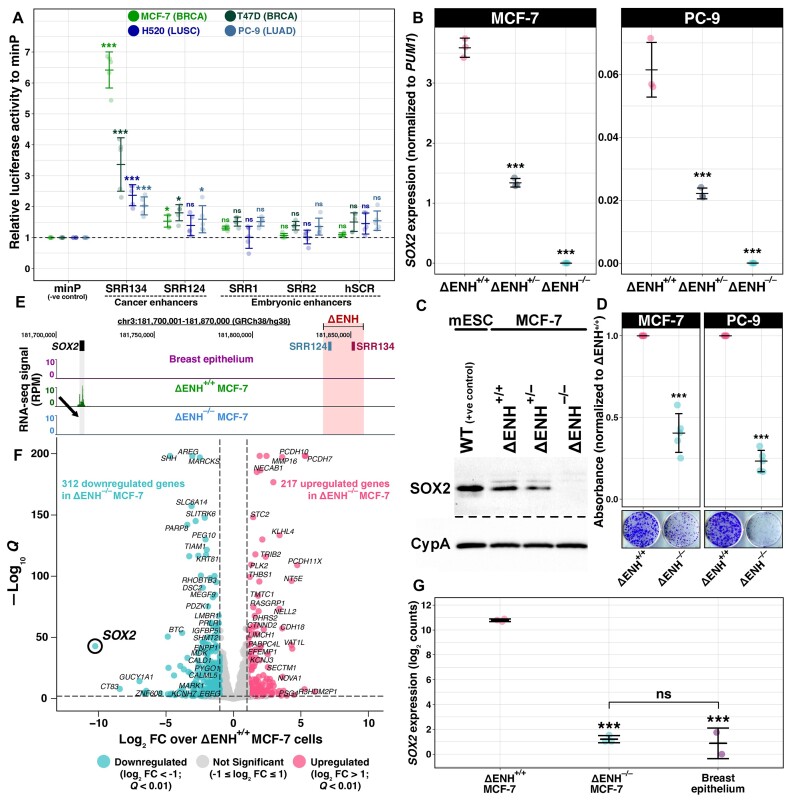
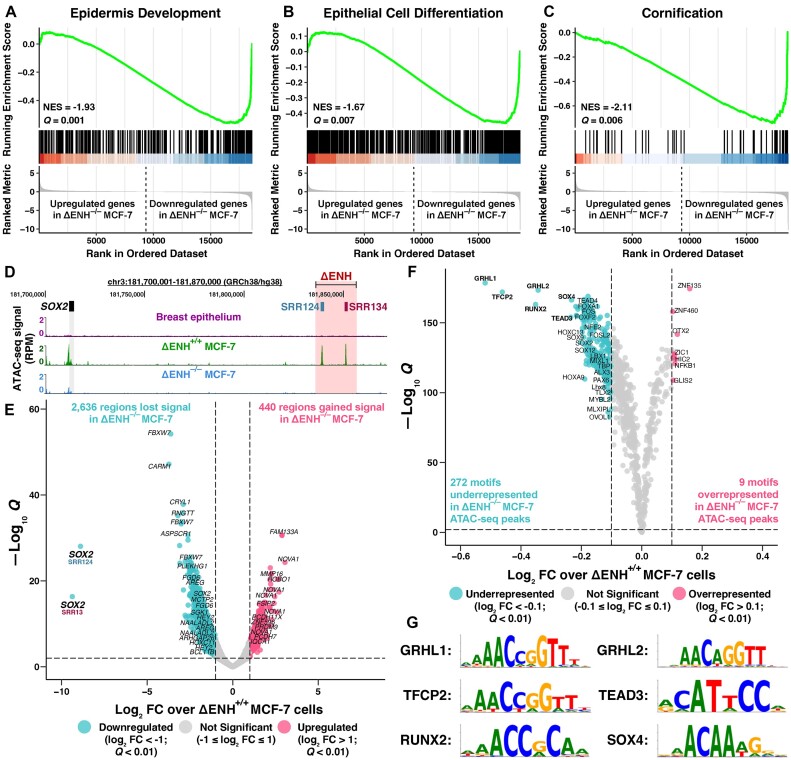
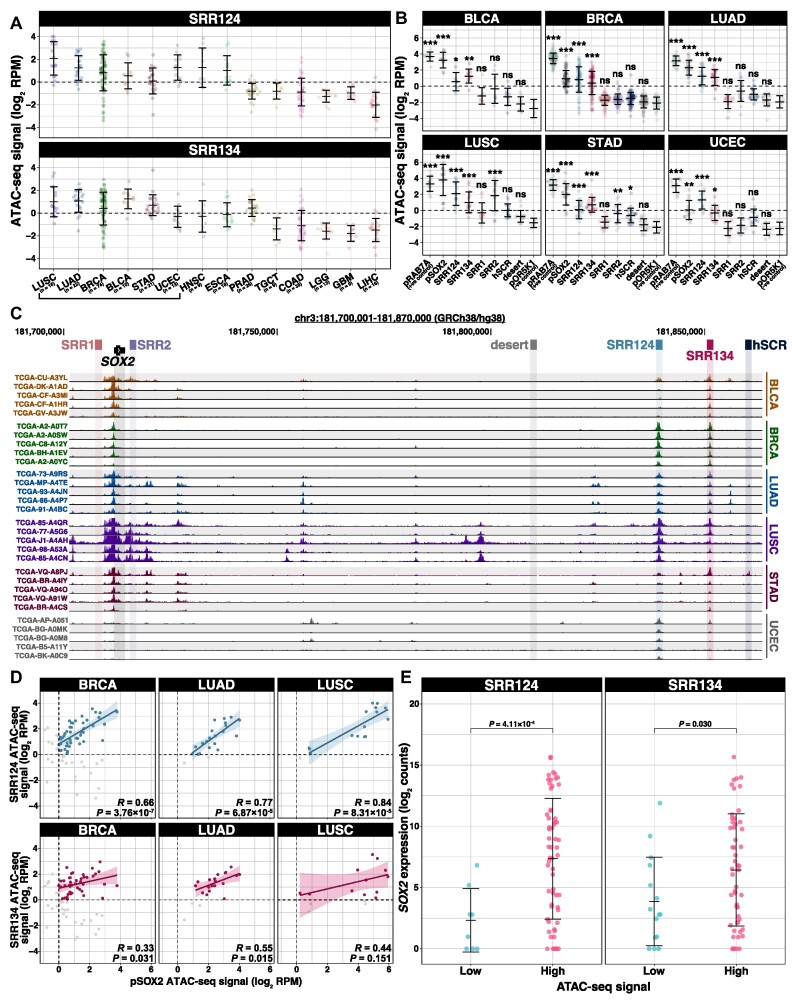
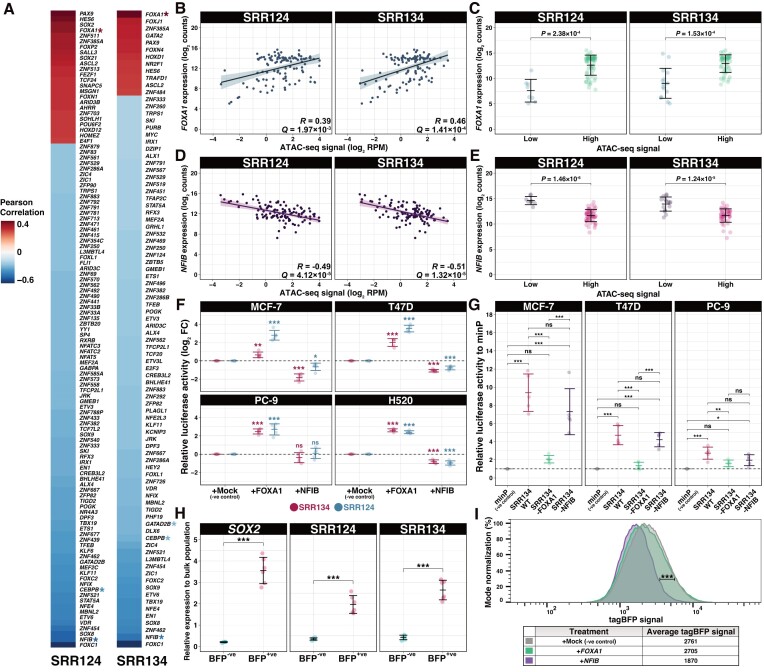
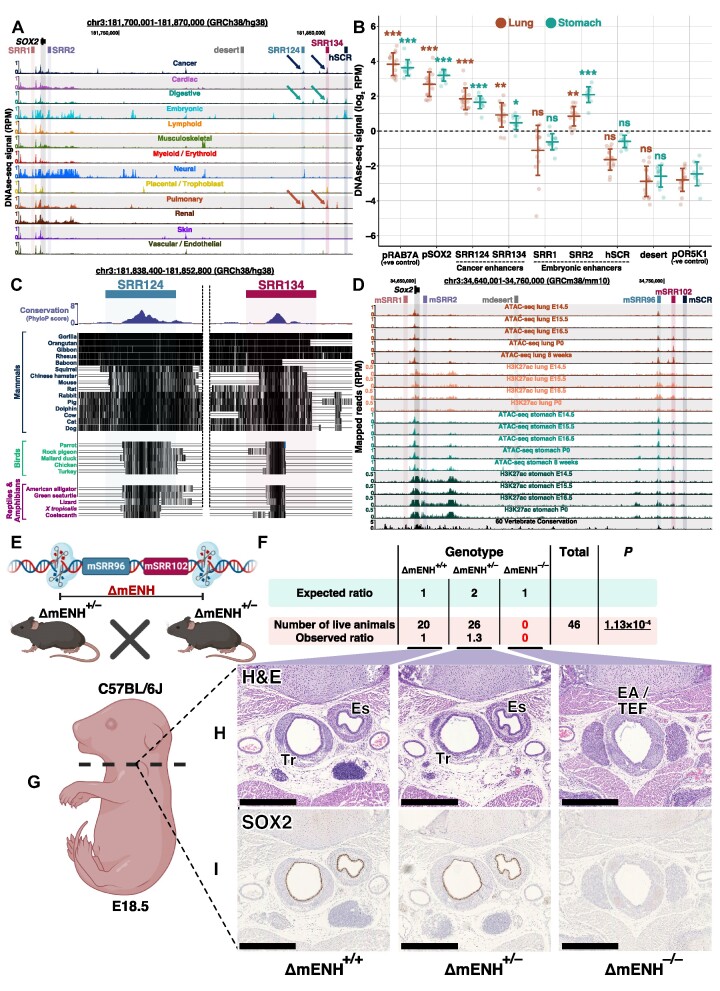
Enhancer reprogramming has been proposed as a key source of transcriptional dysregulation during tumorigenesis, but the molecular mechanisms underlying this process remain unclear. Here, we identify an enhancer cluster required for normal development that is aberrantly activated in breast and lung adenocarcinoma. Deletion of the SRR124-134 cluster disrupts expression of the SOX2 oncogene, dysregulates genome-wide transcription and chromatin accessibility and reduces the ability of cancer cells to form colonies in vitro. Analysis of primary tumors reveals a correlation between chromatin accessibility at this cluster and SOX2 overexpression in breast and lung cancer patients. We demonstrate that FOXA1 is an activator and NFIB is a repressor of SRR124-134 activity and SOX2 transcription in cancer cells, revealing a co-opting of the regulatory mechanisms involved in early development. Notably, we show that the conserved SRR124 and SRR134 regions are essential during mouse development, where homozygous deletion results in the lethal failure of esophageal-tracheal separation. These findings provide insights into how developmental enhancers can be reprogrammed during tumorigenesis and underscore the importance of understanding enhancer dynamics during development and disease.
Plain language summary
The manuscript by Abatti et al. shows that epigenetic reactivation of a pair of distal enhancers that drive Sox2 expression during development (to permit separation of the esophagus and trachea) is responsible for the tumor-promoting re-expression of SOX2 in breast and lung tumors. Intriguingly, the same transcription factors that act on the enhancers during development to either activate or repress them (i.e. FOXA1 and NFIB, respectively) are also required for altering chromatin accessibility of the enhancers and SOX2 transcription in breast and lung cancer cells. With their work, the authors unravel the exact mechanism of how developmentally active enhancers become repurposed in a tumor context and show the relevance of this repurposing event for cancer.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
