

Epigenomic analysis of formalin-fixed paraffin-embedded samples by CUT&Tag
- PMID: 37739938
- PMCID: PMC10516967
- DOI: 10.1038/s41467-023-41666-z
Epigenomic analysis of formalin-fixed paraffin-embedded samples by CUT&Tag
Abstract
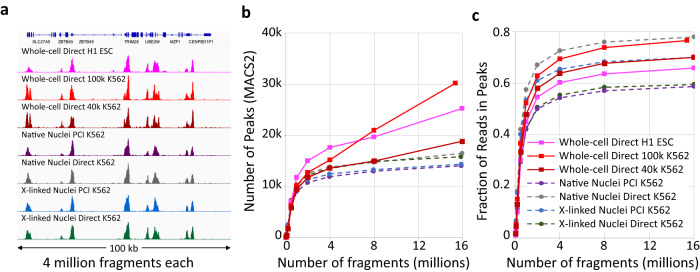
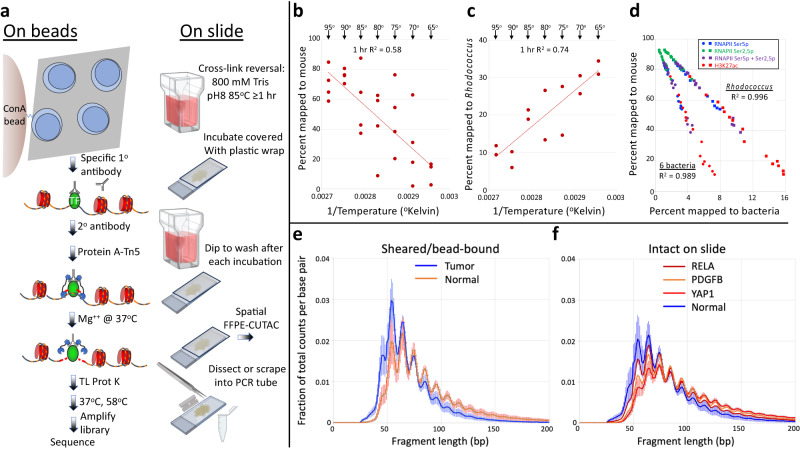
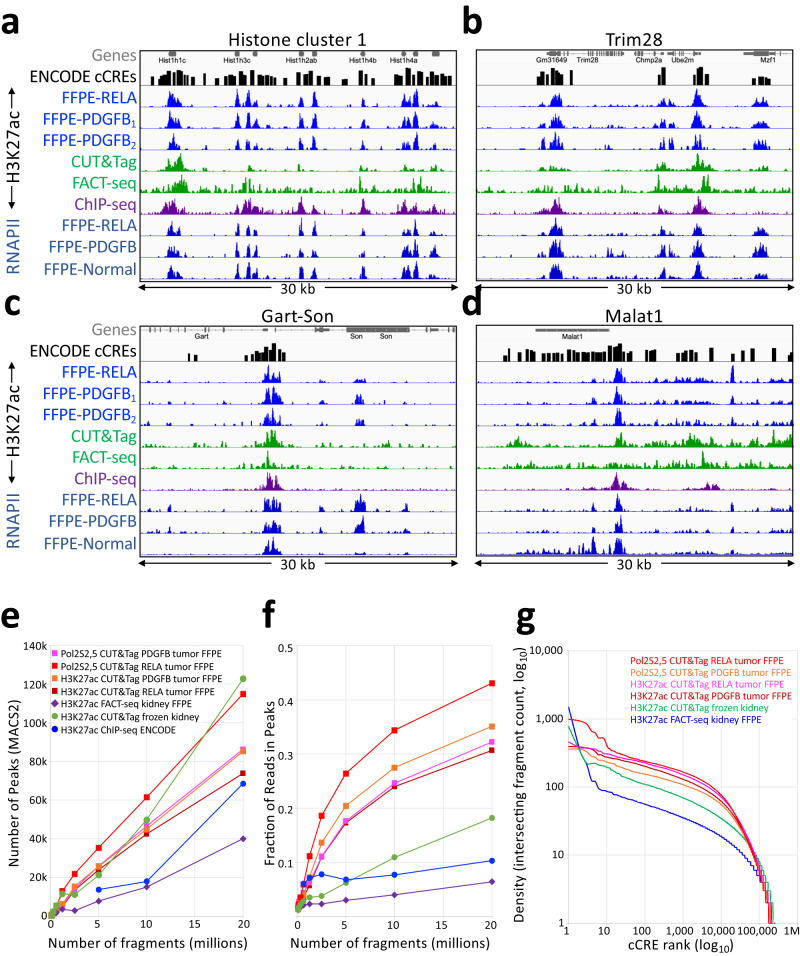
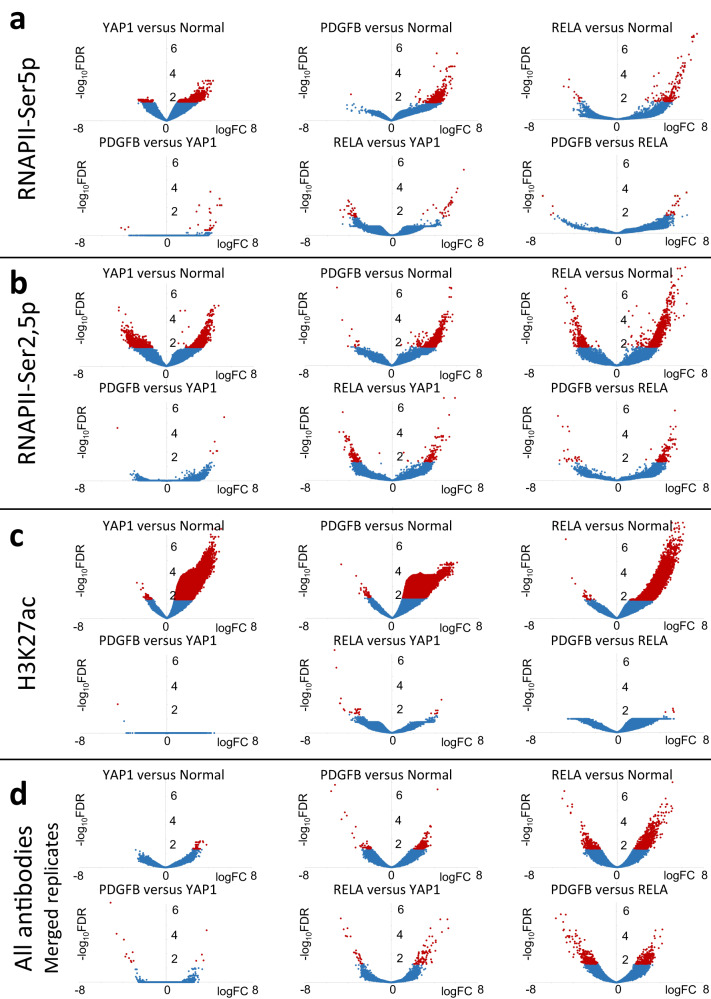
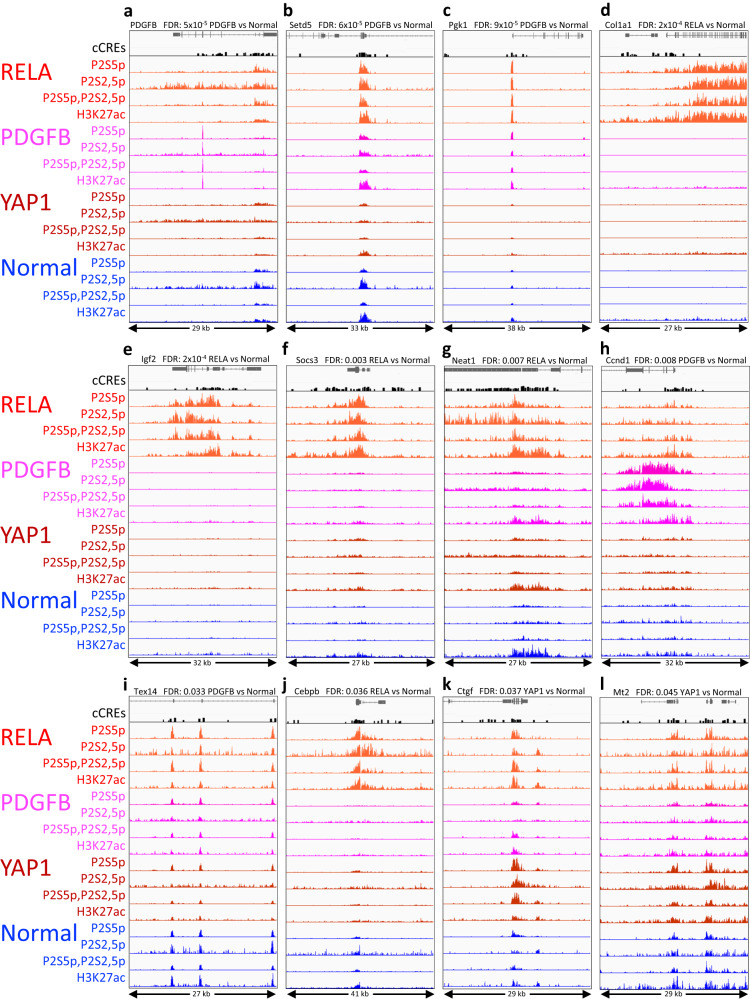
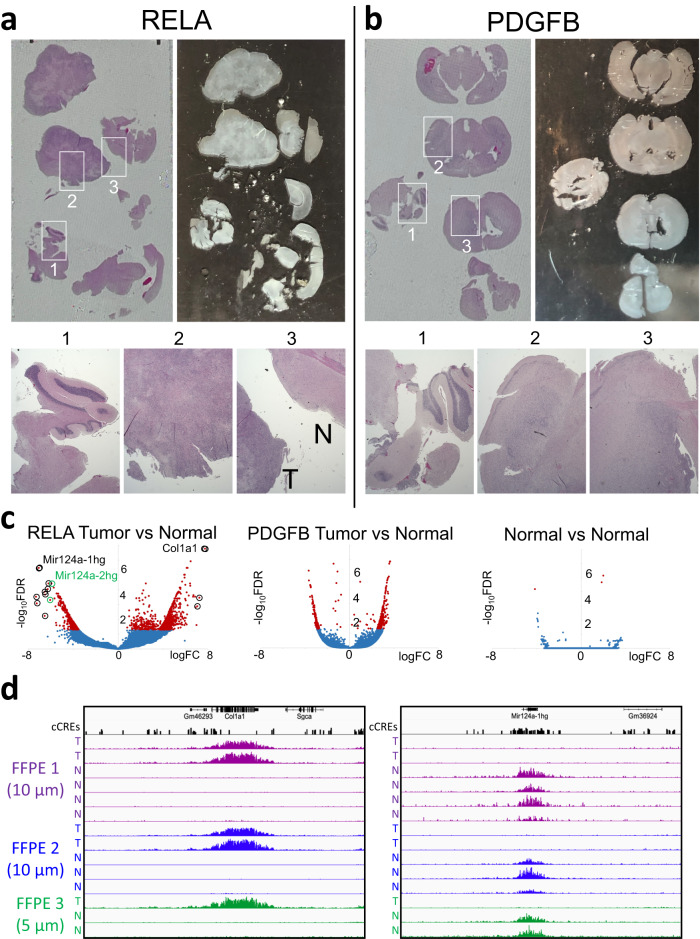
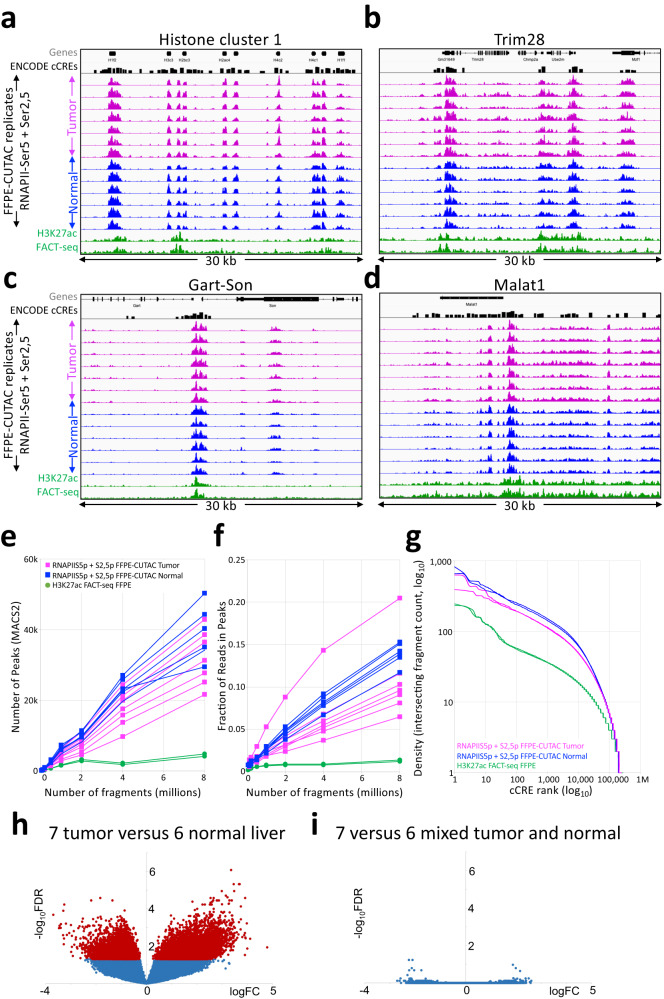
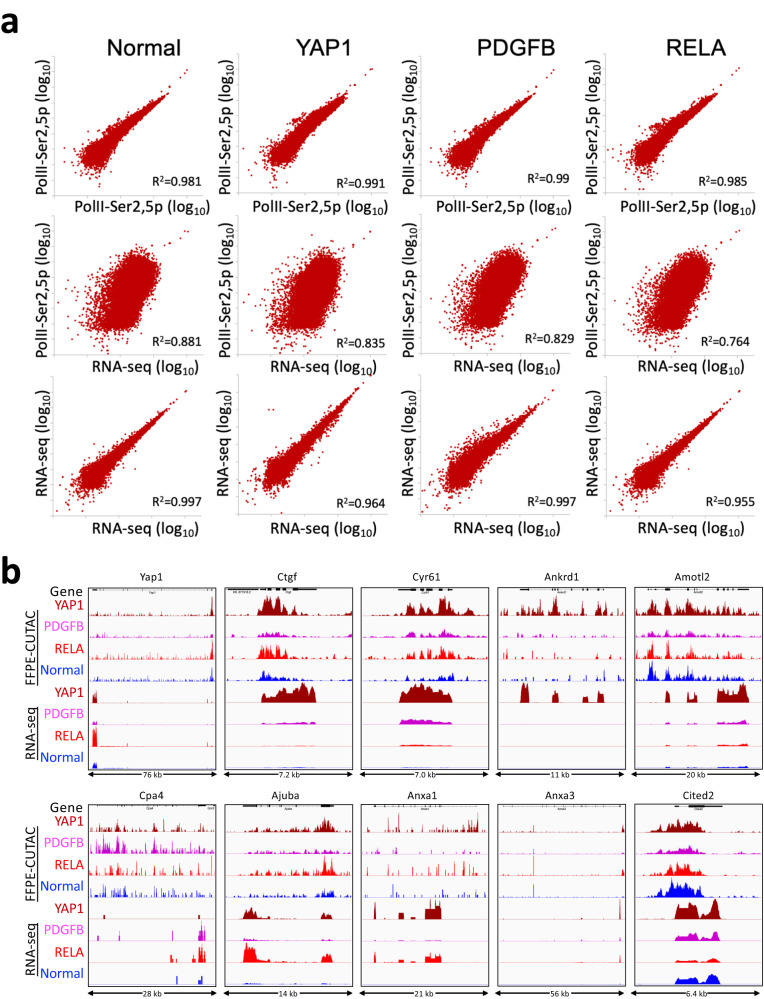
For more than a century, formalin-fixed paraffin-embedded (FFPE) sample preparation has been the preferred method for long-term preservation of biological material. However, the use of FFPE samples for epigenomic studies has been difficult because of chromatin damage from long exposure to high concentrations of formaldehyde. Previously, we introduced Cleavage Under Targeted Accessible Chromatin (CUTAC), an antibody-targeted chromatin accessibility mapping protocol based on CUT&Tag. Here we show that simple modifications of our CUTAC protocol either in single tubes or directly on slides produce high-resolution maps of paused RNA Polymerase II at enhancers and promoters using FFPE samples. We find that transcriptional regulatory element differences produced by FFPE-CUTAC distinguish between mouse brain tumors and identify and map regulatory element markers with high confidence and precision, including microRNAs not detectable by RNA-seq. Our simple workflows make possible affordable epigenomic profiling of archived biological samples for biomarker identification, clinical applications and retrospective studies.
© 2023. Springer Nature Limited.
Conflict of interest statement
S.H. is an inventor in a USPTO patent application filed by the Fred Hutchinson Cancer Center pertaining to CUTAC and FFPE-CUTAC (application number 63/505,964). The other authors declare no competing interests.
Figures
References
-
- Saqi A. The state of cell blocks and ancillary testing: past, present, and future. Arch. Pathol. Lab Med. 2016;140:1318–1322. - PubMed
-
- Blow N. Tissue preparation: tissue issues. Nature. 2007;448:959–963. - PubMed
-
- Armstrong, S. A., Henikoff, S. & Vakoc, C. R. Chromatin Deregulation in Cancer (Cold Spring Harbor Press, 2017).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
