

capTEs enables locus-specific dissection of transcriptional outputs from reference and nonreference transposable elements
- PMID: 37741908
- PMCID: PMC10517987
- DOI: 10.1038/s42003-023-05349-1
capTEs enables locus-specific dissection of transcriptional outputs from reference and nonreference transposable elements
Abstract
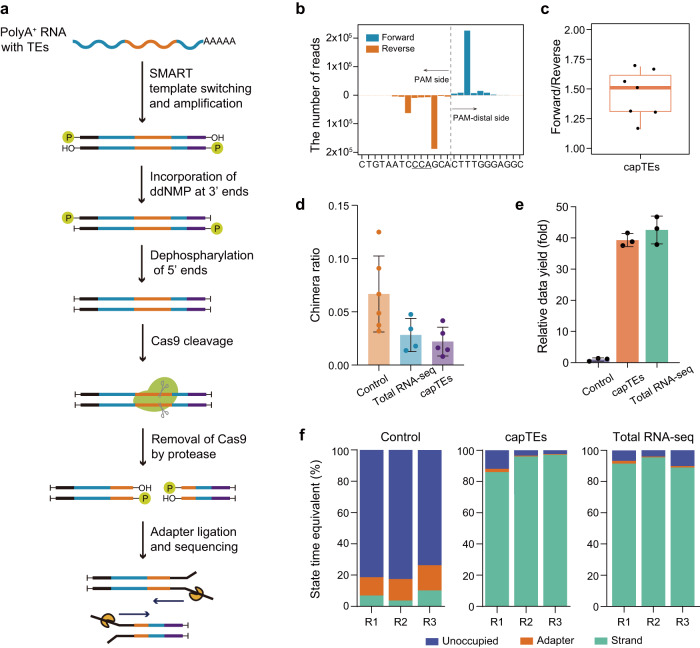
Transposable elements (TEs) serve as both insertional mutagens and regulatory elements in cells, and their aberrant activity is increasingly being revealed to contribute to diseases and cancers. However, measuring the transcriptional consequences of nonreference and young TEs at individual loci remains challenging with current methods, primarily due to technical limitations, including short read lengths generated and insufficient coverage in target regions. Here, we introduce a long-read targeted RNA sequencing method, Cas9-assisted profiling TE expression sequencing (capTEs), for quantitative analysis of transcriptional outputs for individual TEs, including transcribed nonreference insertions, noncanonical transcripts from various transcription patterns and their correlations with expression changes in related genes. This method selectively identified TE-containing transcripts and outputted data with up to 90% TE reads, maintaining a comparable data yield to whole-transcriptome sequencing. We applied capTEs to human cancer cells and found that internal and inserted Alu elements may employ distinct regulatory mechanisms to upregulate gene expression. We expect that capTEs will be a critical tool for advancing our understanding of the biological functions of individual TEs at the locus level, revealing their roles as both mutagens and regulators in biological and pathogenic processes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
