

Iterative machine learning-based chemical similarity search to identify novel chemical inhibitors
- PMID: 37742003
- PMCID: PMC10517535
- DOI: 10.1186/s13321-023-00760-6
Iterative machine learning-based chemical similarity search to identify novel chemical inhibitors
Abstract
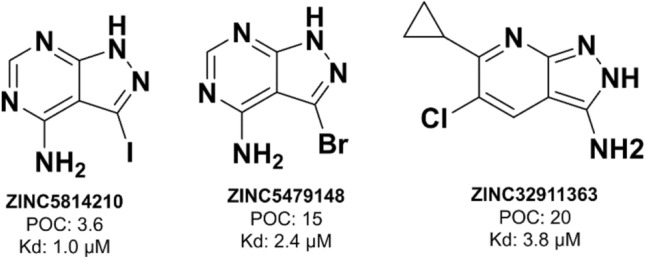
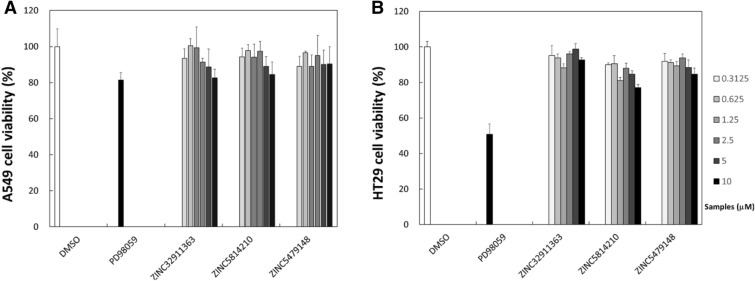
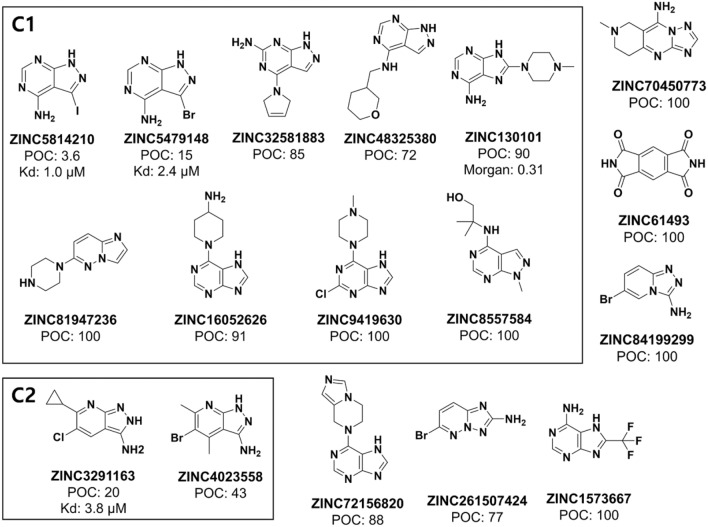
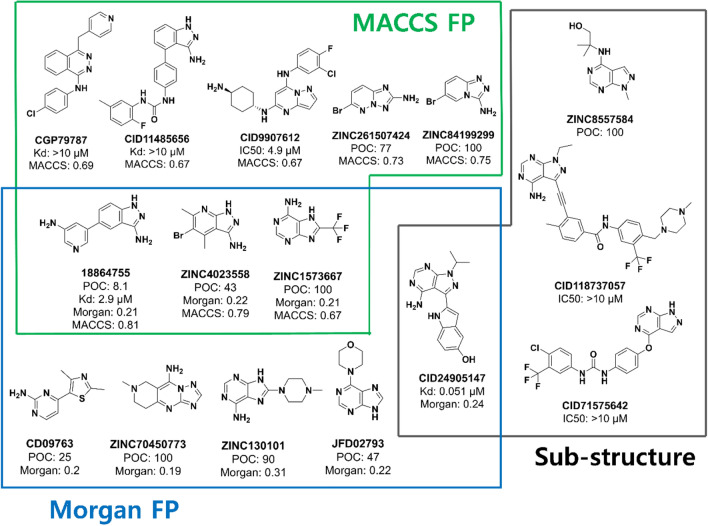
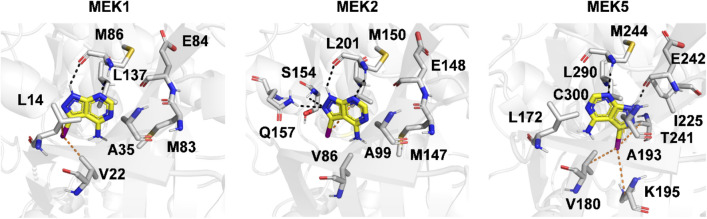
Machine learning-based chemical screening has made substantial progress in recent years. However, these predictions often have low accuracy and high uncertainty when identifying new active chemical scaffolds. Hence, a high proportion of retrieved compounds are not structurally novel. In this study, we proposed a strategy to address this issue by iteratively optimizing an evolutionary chemical binding similarity (ECBS) model using experimental validation data. Various data update and model retraining schemes were tested to efficiently incorporate new experimental data into ECBS models, resulting in a fine-tuned ECBS model with improved accuracy and coverage. To demonstrate the effectiveness of our approach, we identified the novel hit molecules for the mitogen-activated protein kinase kinase 1 (MEK1). These molecules showed sub-micromolar affinity (Kd 0.1-5.3 μM) to MEKs and were distinct from previously-known MEK1 inhibitors. We also determined the binding specificity of different MEK isoforms and proposed potential docking models. Furthermore, using de novo drug design tools, we utilized one of the new MEK inhibitors to generate additional drug-like molecules with improved binding scores. This resulted in the identification of several potential MEK1 inhibitors with better binding affinity scores. Our results demonstrated the potential of this approach for identifying novel hit molecules and optimizing their binding affinities.
Keywords: Drug design; Evolutionary chemical binding similarity; Hit identification; MEK inhibitor; Virtual screening.
© 2023. Springer Nature Switzerland AG.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


Similar articles
-
Evolutionary chemical binding similarity approach integrated with 3D-QSAR method for effective virtual screening.BMC Bioinformatics. 2020 Jul 14;21(1):309. doi: 10.1186/s12859-020-03643-x. BMC Bioinformatics. 2020. PMID: 32664863 Free PMC article.
-
Interpretable Machine Learning Models for Molecular Design of Tyrosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration.Int J Mol Sci. 2022 Sep 24;23(19):11262. doi: 10.3390/ijms231911262. Int J Mol Sci. 2022. PMID: 36232566 Free PMC article.
-
Identifying Novel Inhibitors for Hepatic Organic Anion Transporting Polypeptides by Machine Learning-Based Virtual Screening.J Chem Inf Model. 2022 Dec 26;62(24):6323-6335. doi: 10.1021/acs.jcim.1c01460. Epub 2022 Mar 11. J Chem Inf Model. 2022. PMID: 35274943 Free PMC article. Review.
-
[Docking-based virtual screening of MEK1 inhibitors from Chinese herbs].Zhongguo Zhong Yao Za Zhi. 2017 May;42(10):1951-1956. doi: 10.19540/j.cnki.cjcmm.2017.0083. Zhongguo Zhong Yao Za Zhi. 2017. PMID: 29090556 Chinese.
-
Applications of Quantitative Structure-Activity Relationships (QSAR) based Virtual Screening in Drug Design: A Review.Mini Rev Med Chem. 2020;20(14):1375-1388. doi: 10.2174/1389557520666200429102334. Mini Rev Med Chem. 2020. PMID: 32348219 Review.
Cited by
-
Supervised Screening of EGFR Inhibitors Validated through Computational Structural Biology Approaches.ACS Med Chem Lett. 2024 Dec 2;15(12):2190-2200. doi: 10.1021/acsmedchemlett.4c00385. eCollection 2024 Dec 12. ACS Med Chem Lett. 2024. PMID: 39691517
-
Optimization of separation and purification processes in diethyl ether production for improved efficiency and sustainability.Sci Rep. 2025 Jul 31;15(1):27911. doi: 10.1038/s41598-025-10516-x. Sci Rep. 2025. PMID: 40744968 Free PMC article.
References
-
- Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, Terentiev VA, Polykovskiy DA, Kuznetsov MD, Asadulaev A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019;37(9):1038–1040. doi: 10.1038/s41587-019-0224-x. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
