

Vepafestinib is a pharmacologically advanced RET-selective inhibitor with high CNS penetration and inhibitory activity against RET solvent front mutations
- PMID: 37743366
- PMCID: PMC10518257
- DOI: 10.1038/s43018-023-00630-y
Vepafestinib is a pharmacologically advanced RET-selective inhibitor with high CNS penetration and inhibitory activity against RET solvent front mutations
Erratum in
-
Author Correction: Vepafestinib is a pharmacologically advanced RET-selective inhibitor with high CNS penetration and inhibitory activity against RET solvent front mutations.Nat Cancer. 2023 Oct;4(10):1526. doi: 10.1038/s43018-023-00663-3. Nat Cancer. 2023. PMID: 37814012 Free PMC article. No abstract available.
Abstract
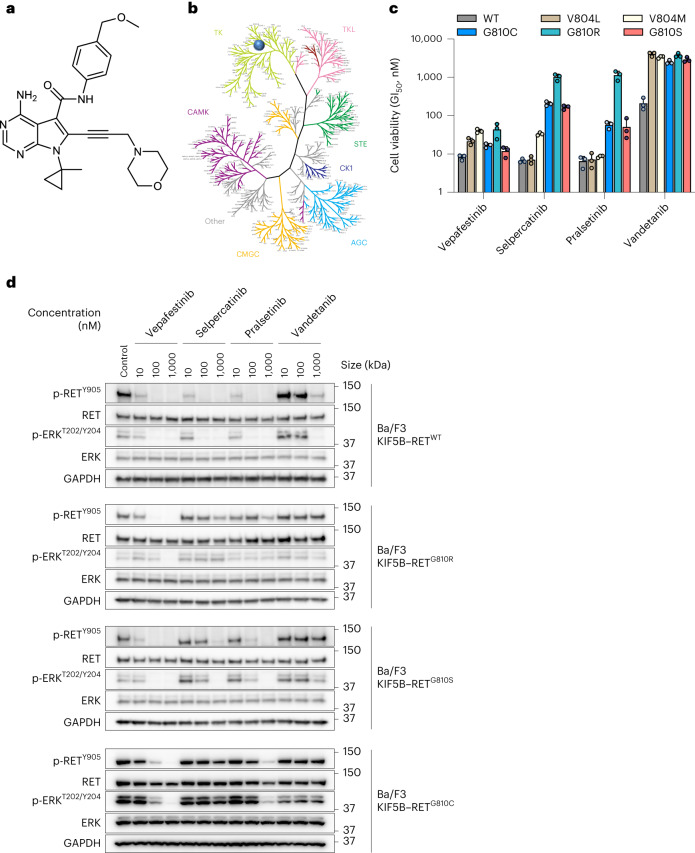
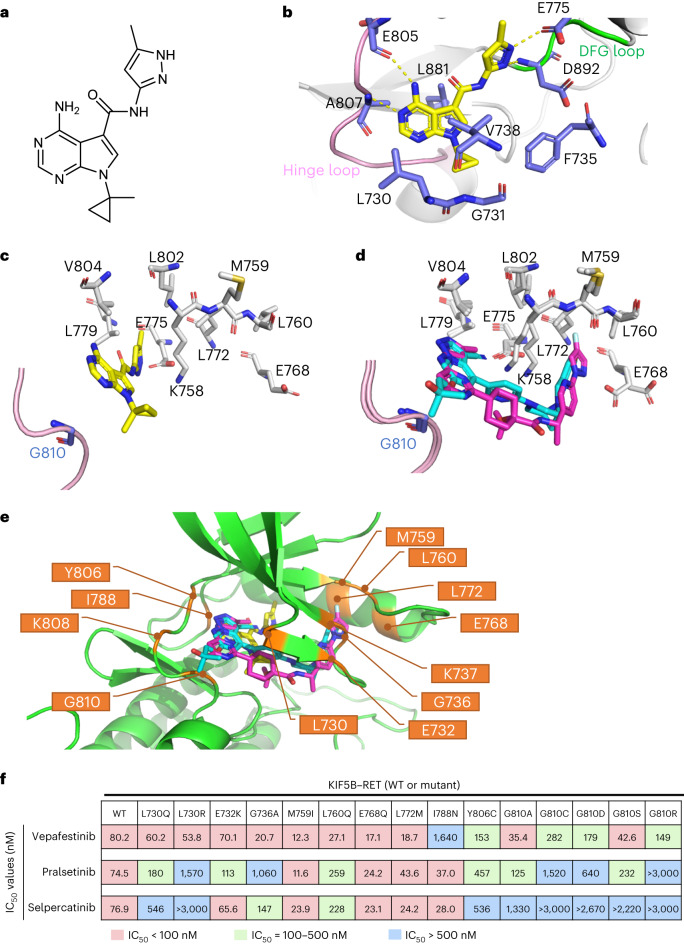
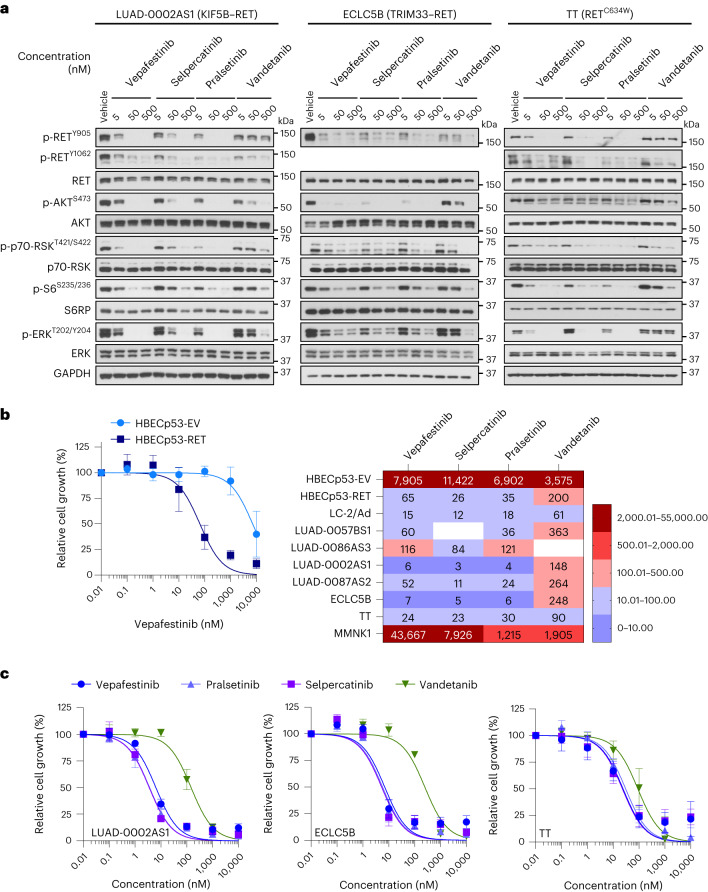
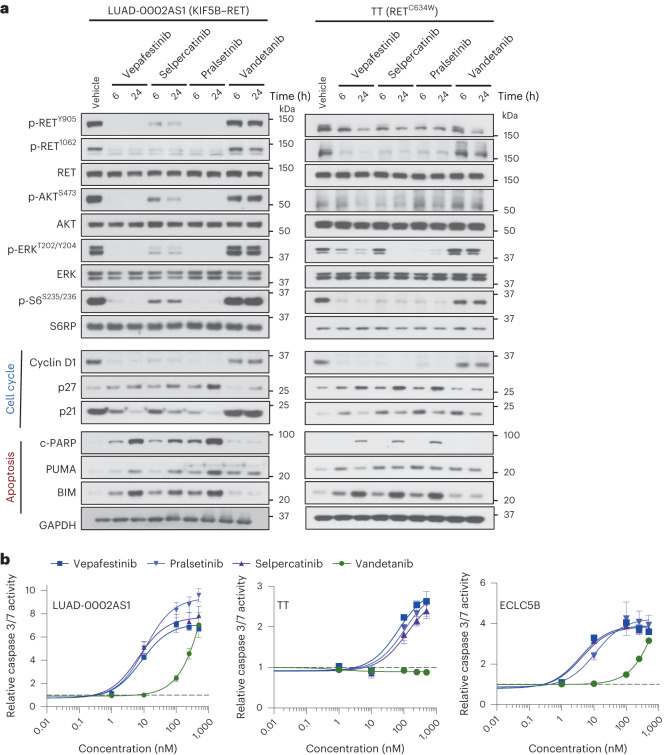
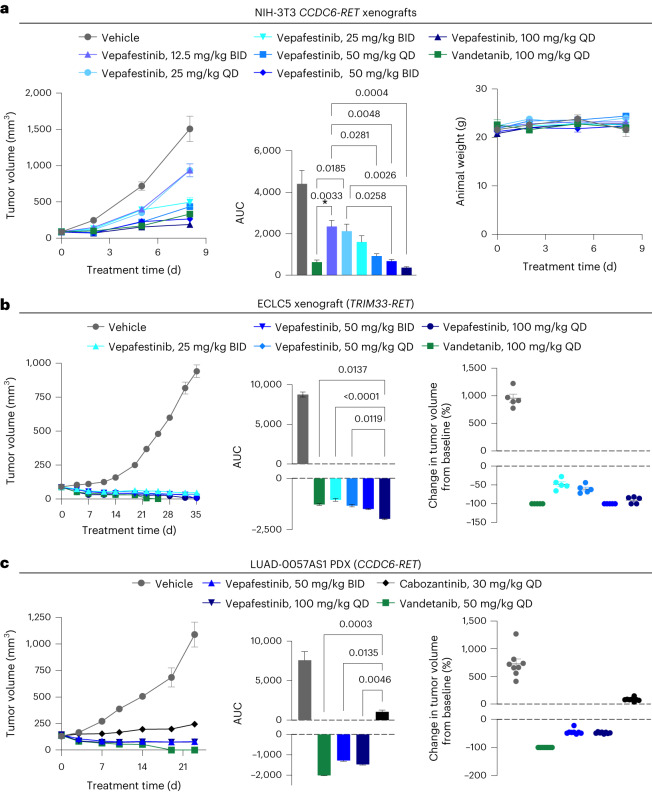
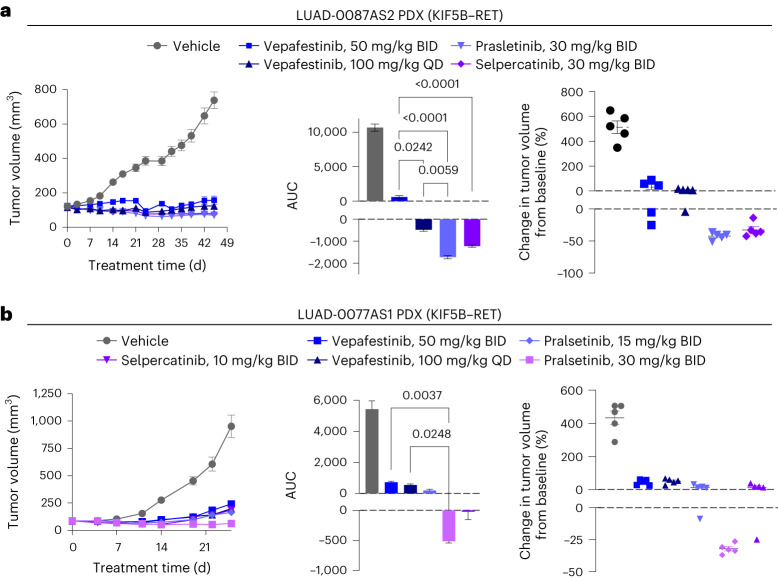
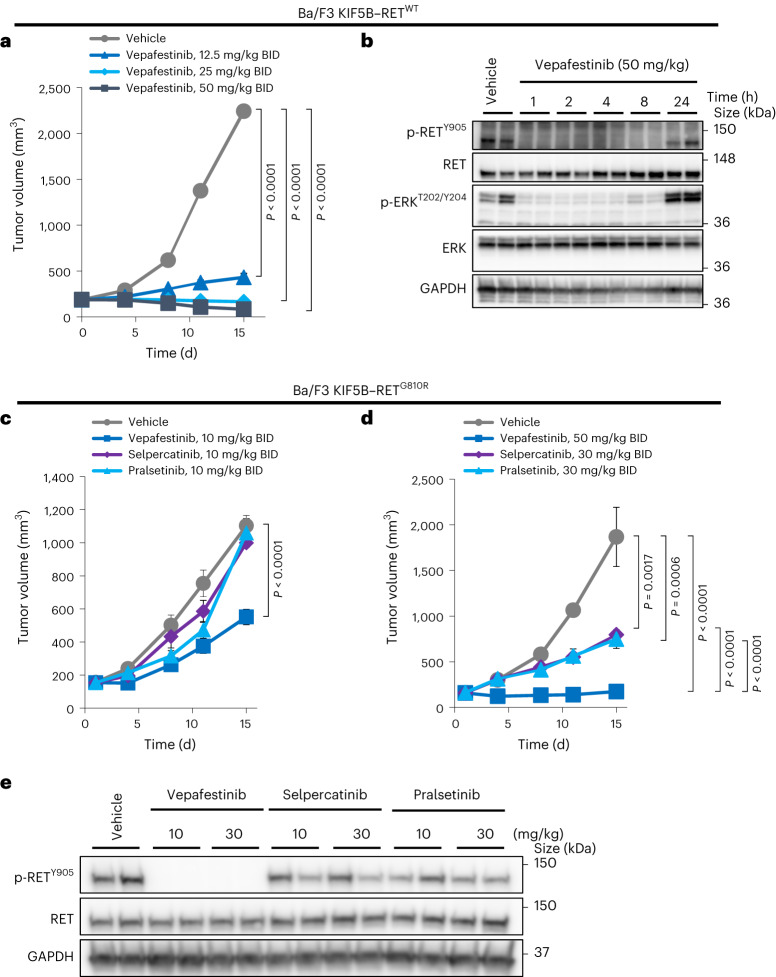
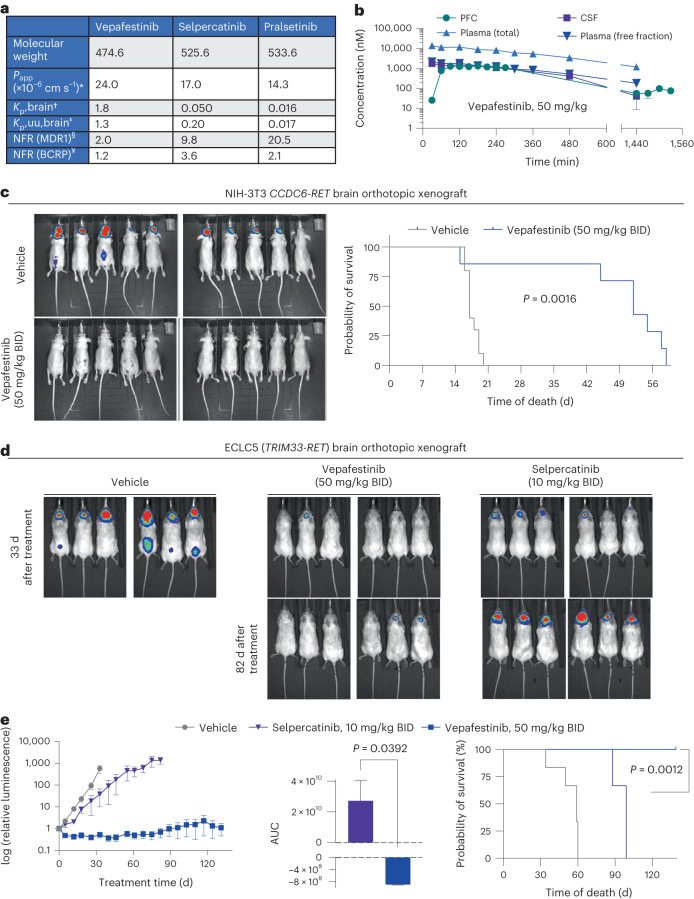
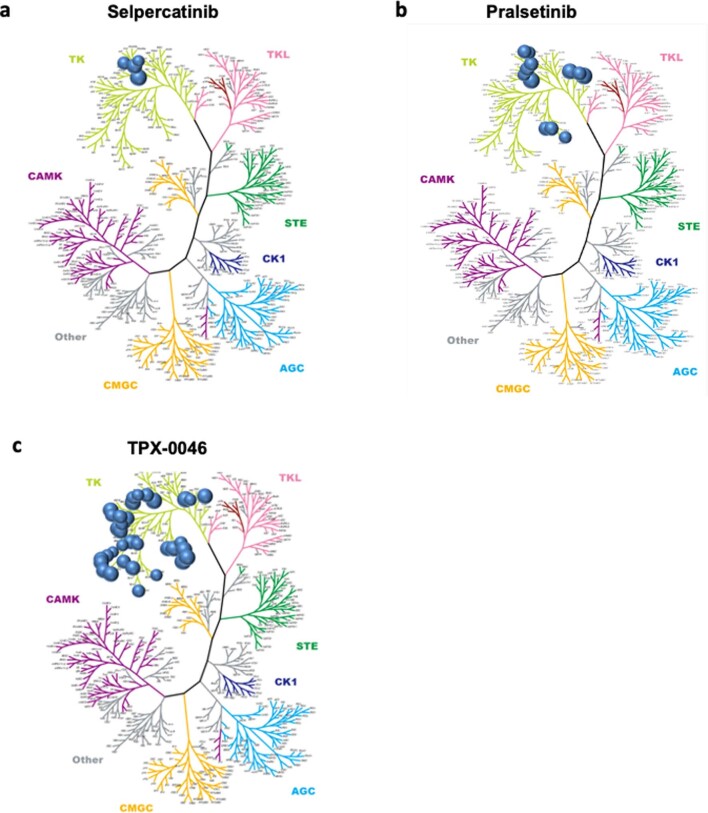
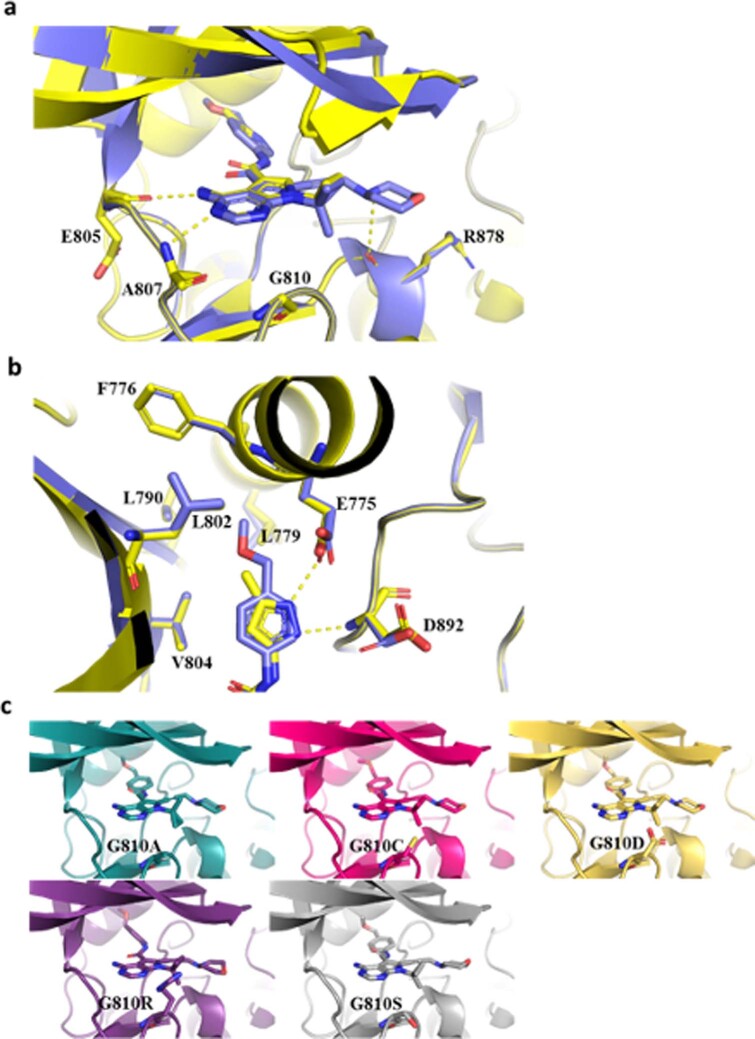
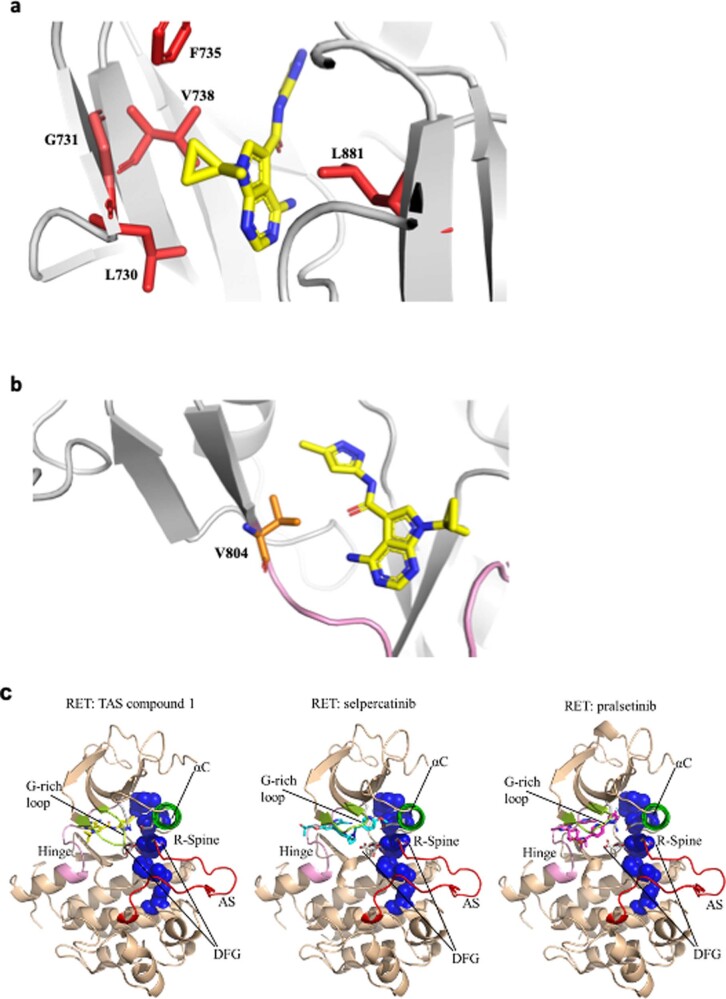
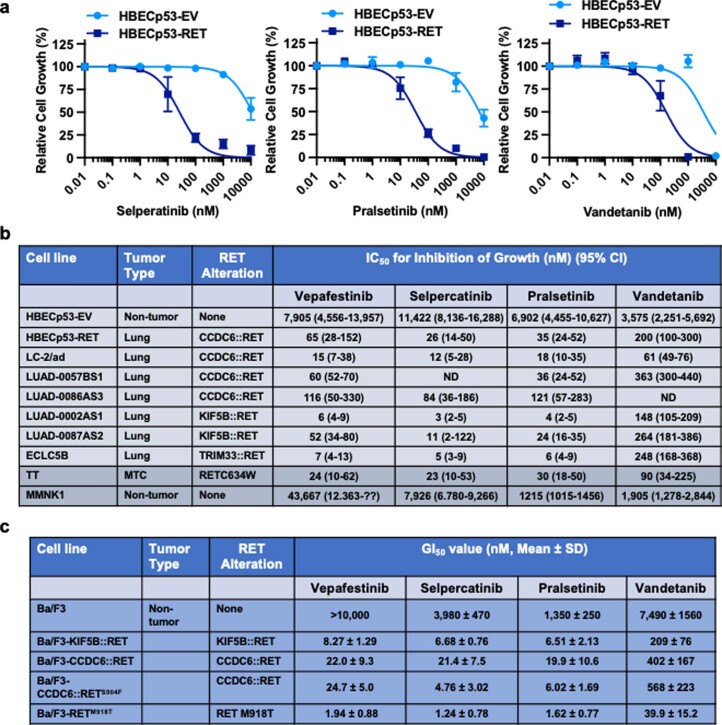
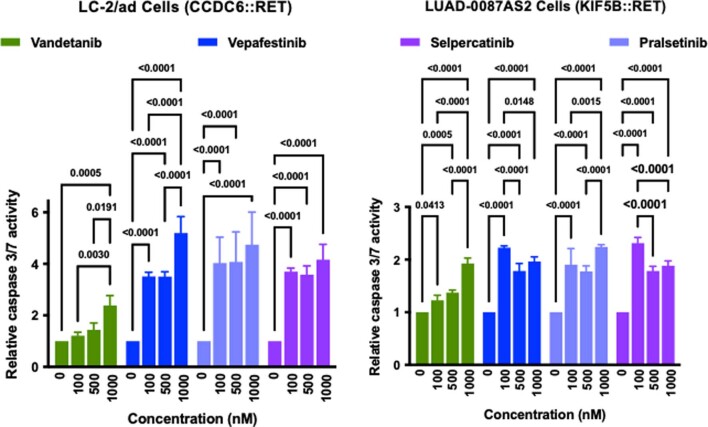
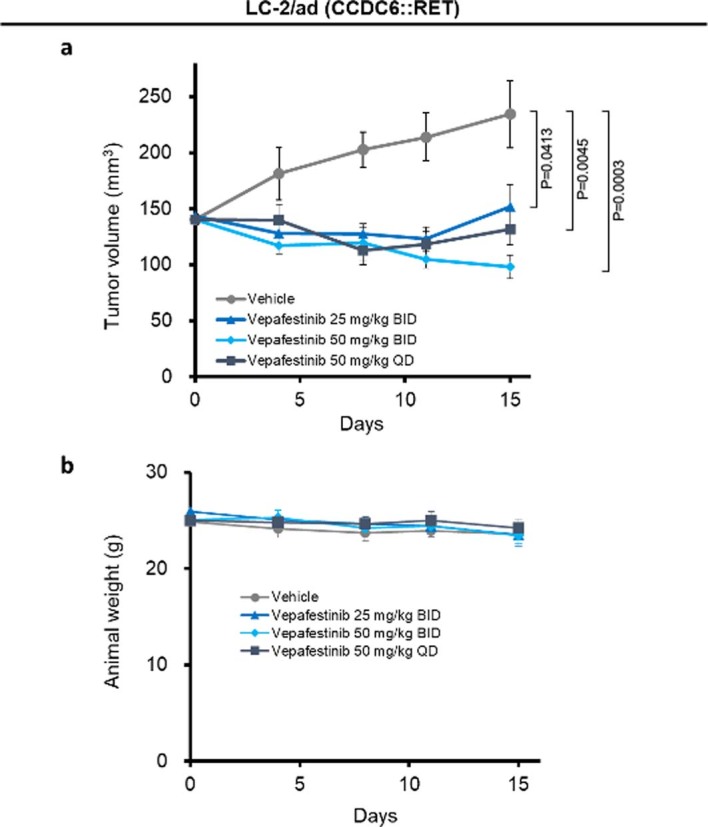
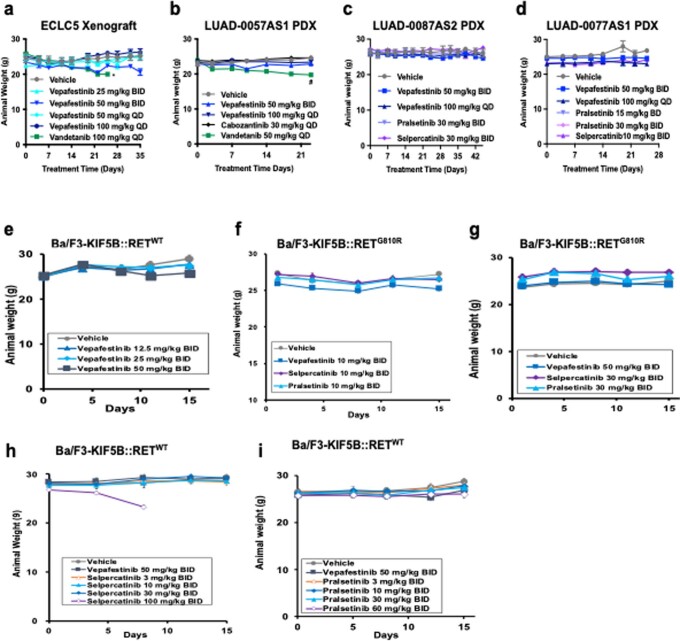
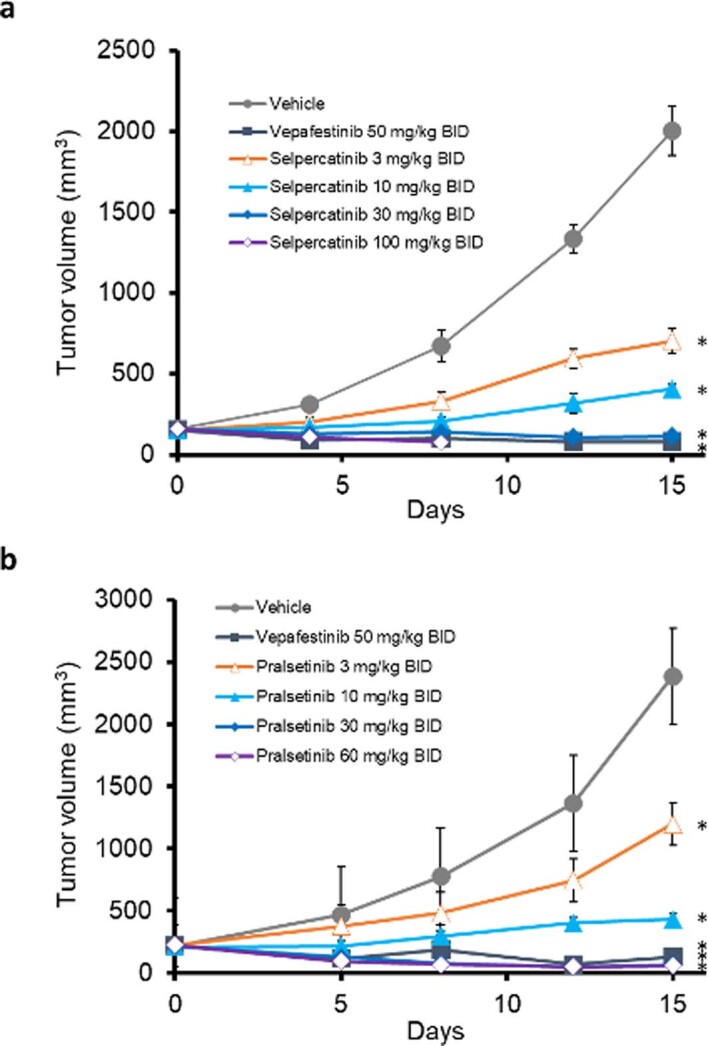
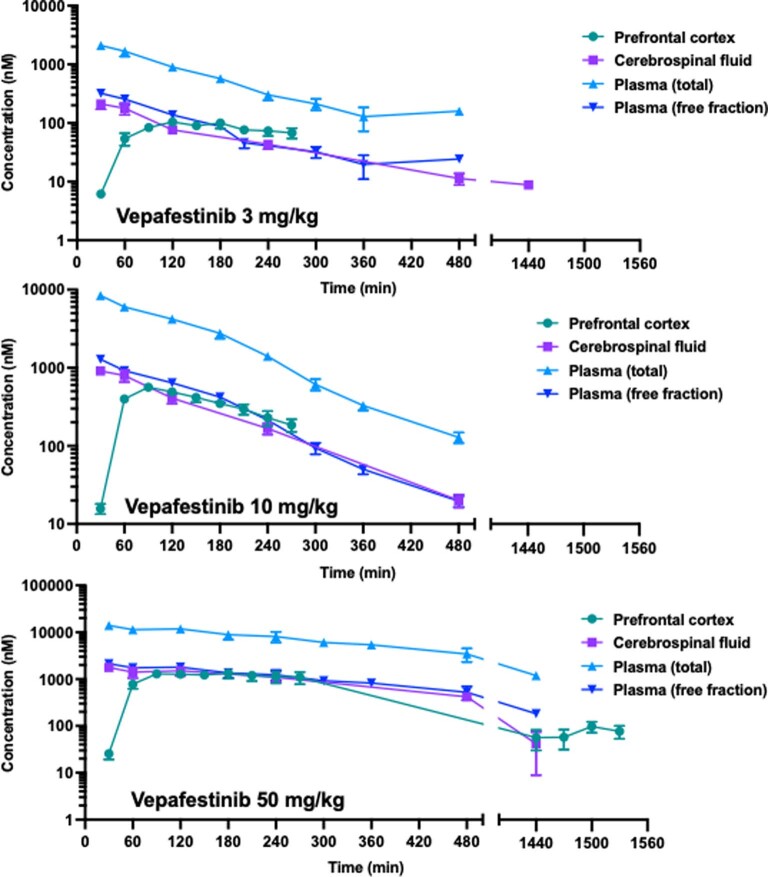
RET receptor tyrosine kinase is activated in various cancers (lung, thyroid, colon and pancreatic, among others) through oncogenic fusions or gain-of-function single-nucleotide variants. Small-molecule RET kinase inhibitors became standard-of-care therapy for advanced malignancies driven by RET. The therapeutic benefit of RET inhibitors is limited, however, by acquired mutations in the drug target as well as brain metastasis, presumably due to inadequate brain penetration. Here, we perform preclinical characterization of vepafestinib (TAS0953/HM06), a next-generation RET inhibitor with a unique binding mode. We demonstrate that vepafestinib has best-in-class selectivity against RET, while exerting activity against commonly reported on-target resistance mutations (variants in RETL730, RETV804 and RETG810), and shows superior pharmacokinetic properties in the brain when compared to currently approved RET drugs. We further show that these properties translate into improved tumor control in an intracranial model of RET-driven cancer. Our results underscore the clinical potential of vepafestinib in treating RET-driven cancers.
© 2023. The Author(s).
Conflict of interest statement
I.O., A.J.W.L., L.D., I.K., M.S.M., K. Ishizawa, M.V., Y.C.L., R.C., T.Z., R.I.K., S.K., Q.C., E.C., E.d.S. and M.A.D. report no potential conflict of interest. M.L. has received advisory board compensation from Merck, Bristol Myers Squibb, Takeda, Bayer, Lilly Oncology, Janssen and Paige.AI. In addition, research grants unrelated to the current study were obtained from Merus, Loxo Oncology and Elevation Oncology. Research funding from Helsinn Healthcare partially supported this study. R.S. has received research support from Merus, Loxo Oncology and Elevation Oncology, unrelated to the current study. I.M., K. Ishida, M.K., T.S., K.W., H.F., Y.Y., K.F., S.O., W.Y., R.T. and Y.I. are employees of Taiho Pharmaceutical. E.L., C.G. and A.B. are employees of Helsinn Healthcare.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
