

Non-small cell lung cancers (NSCLCs) oncolysis using coxsackievirus B5 and synergistic DNA-damage response inhibitors
- PMID: 37743418
- PMCID: PMC10518312
- DOI: 10.1038/s41392-023-01603-4
Non-small cell lung cancers (NSCLCs) oncolysis using coxsackievirus B5 and synergistic DNA-damage response inhibitors
Abstract
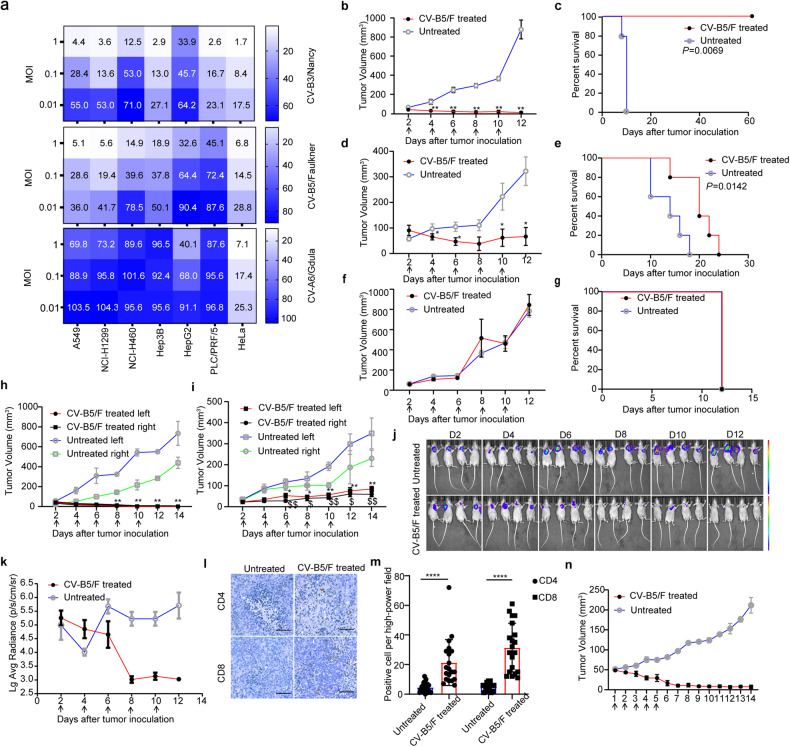
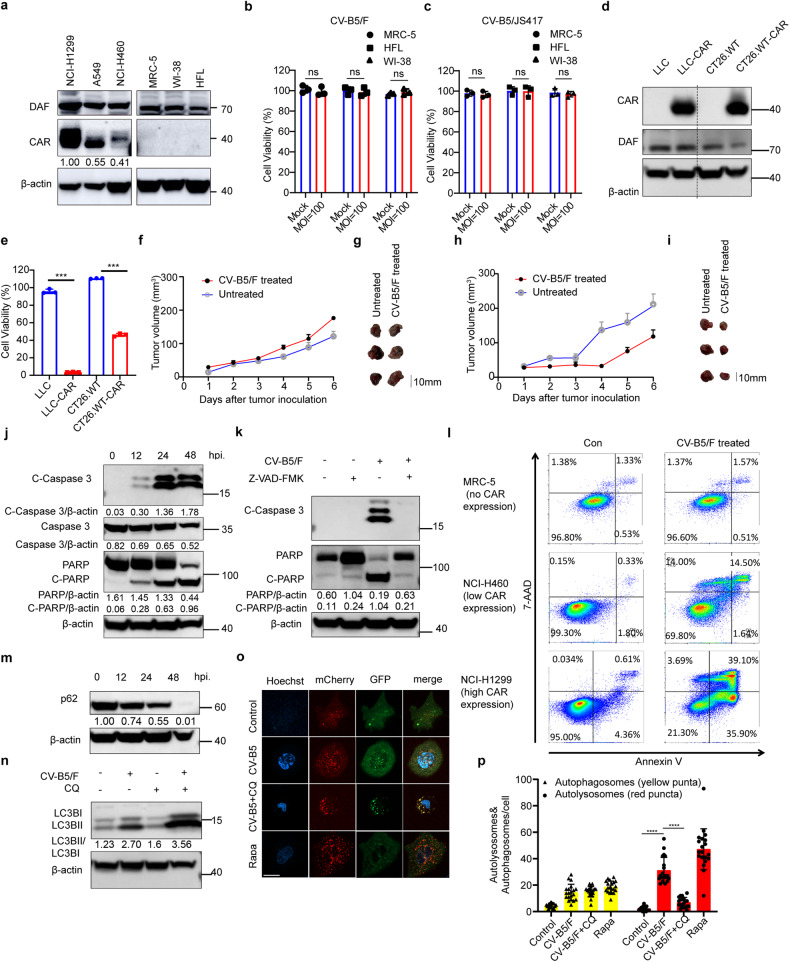
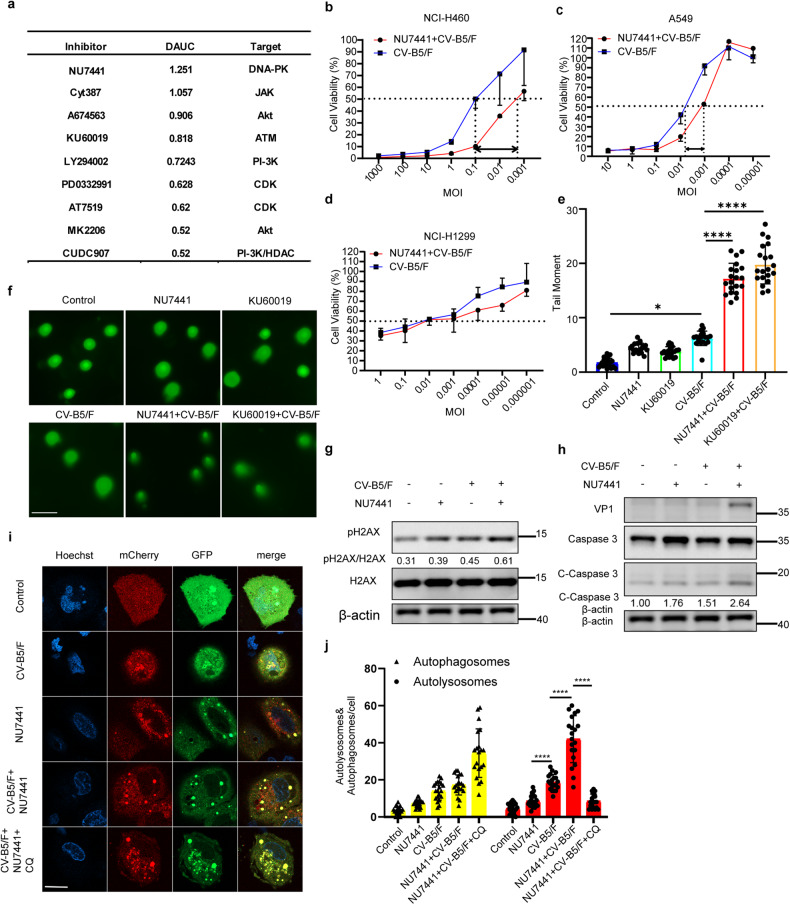
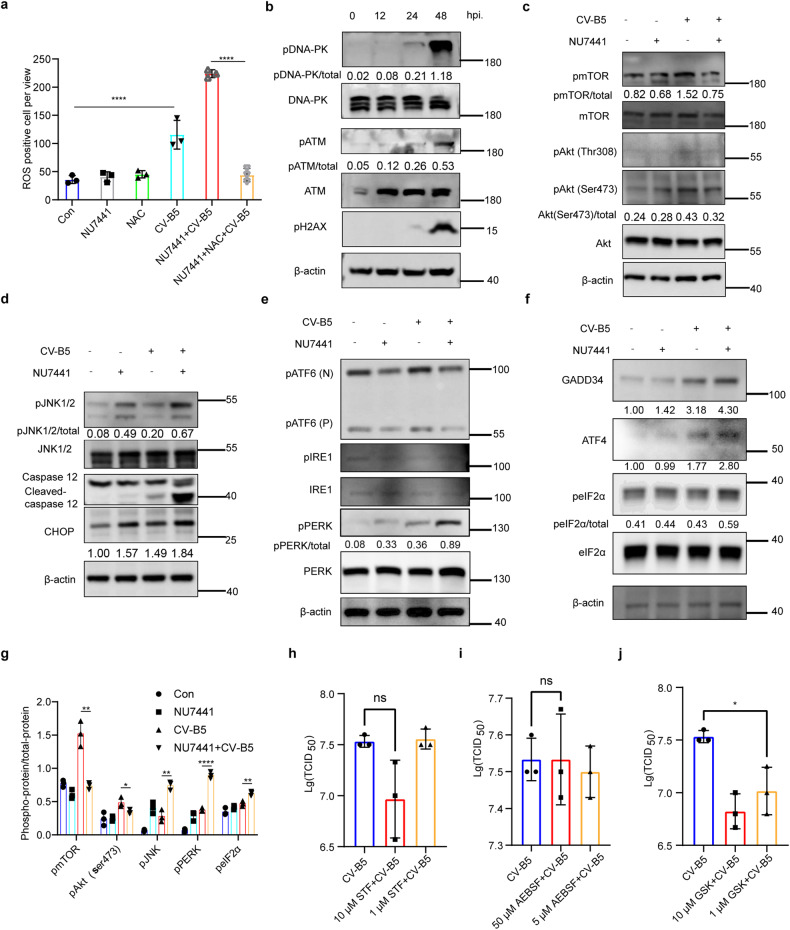
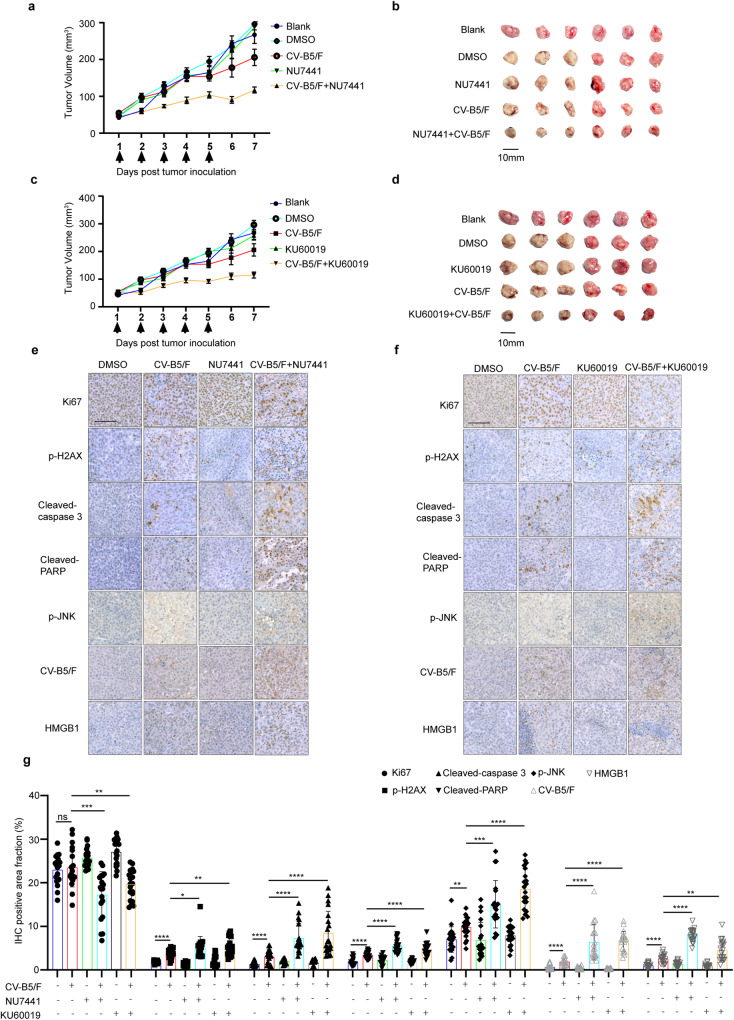
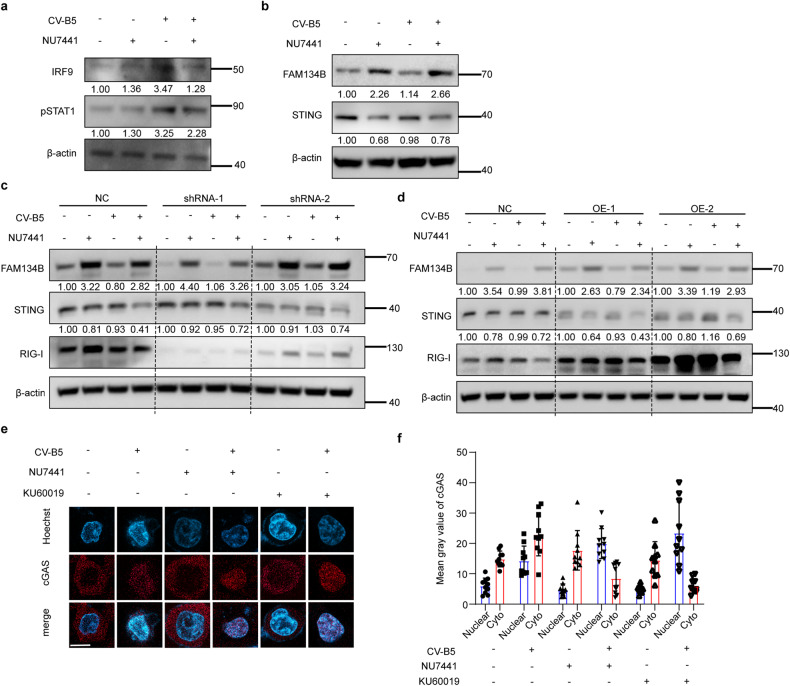
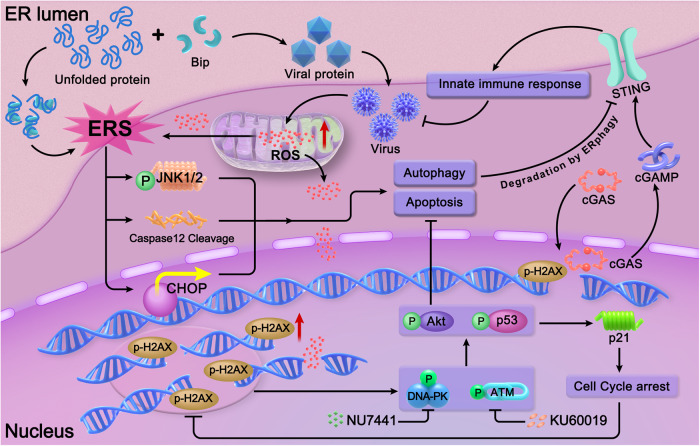
With the continuous in-depth study of the interaction mechanism between viruses and hosts, the virus has become a promising tool in cancer treatment. In fact, many oncolytic viruses with selectivity and effectiveness have been used in cancer therapy. Human enterovirus is one of the most convenient sources to generate oncolytic viruses, however, the high seroprevalence of some enteroviruses limits its application which urges to exploit more oncolytic enteroviruses. In this study, coxsackievirus B5/Faulkner (CV-B5/F) was screened for its potential oncolytic effect against non-small cell lung cancers (NSCLCs) through inducing apoptosis and autophagy. For refractory NSCLCs, DNA-dependent protein kinase (DNA-PK) or ataxia telangiectasia mutated protein (ATM) inhibitors can synergize with CV-B5/F to promote refractory cell death. Here, we showed that viral infection triggered endoplasmic reticulum (ER) stress-related pro-apoptosis and autophagy signals, whereas repair for double-stranded DNA breaks (DSBs) contributed to cell survival which can be antagonized by inhibitor-induced cell death, manifesting exacerbated DSBs, apoptosis, and autophagy. Mechanistically, PERK pathway was activated by the combination of CV-B5/F and inhibitor, and the irreversible ER stress-induced exacerbated cell death. Furthermore, the degradation of activated STING by ERphagy promoted viral replication. Meanwhile, no treatment-related deaths due to CV-B5/F and/or inhibitors occurred. Conclusively, our study identifies an oncolytic CV-B5/F and the synergistic effects of inhibitors of DNA-PK or ATM, which is a potential therapy for NSCLCs.
© 2023. West China Hospital, Sichuan University.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
