

Neuroinflammation, autoinflammation, splenomegaly and anemia caused by bi-allelic mutations in IRAK4
- PMID: 37744344
- PMCID: PMC10516541
- DOI: 10.3389/fimmu.2023.1231749
Neuroinflammation, autoinflammation, splenomegaly and anemia caused by bi-allelic mutations in IRAK4
Abstract
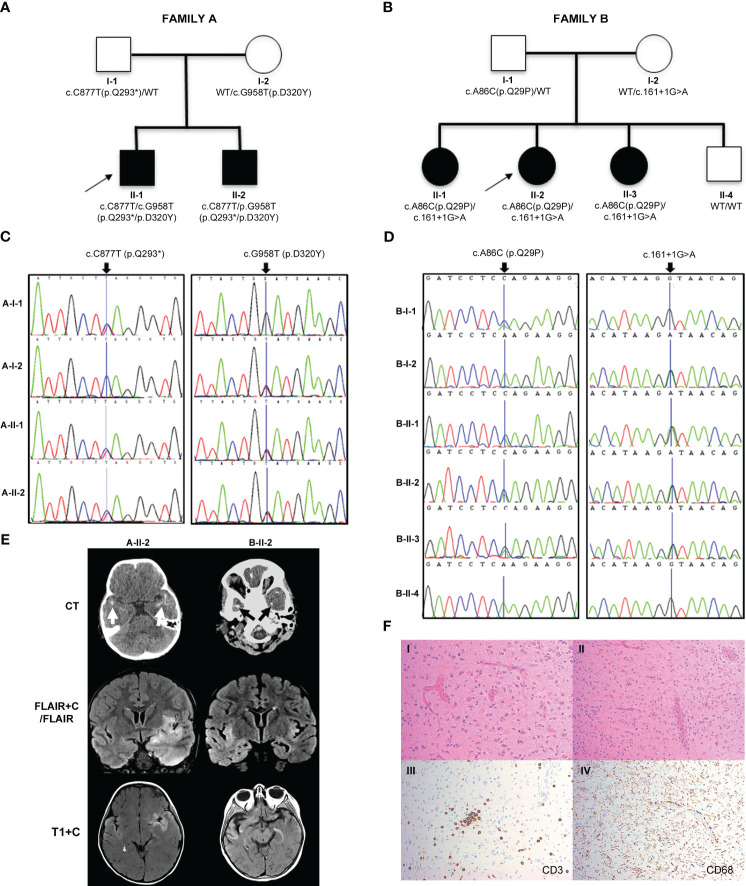
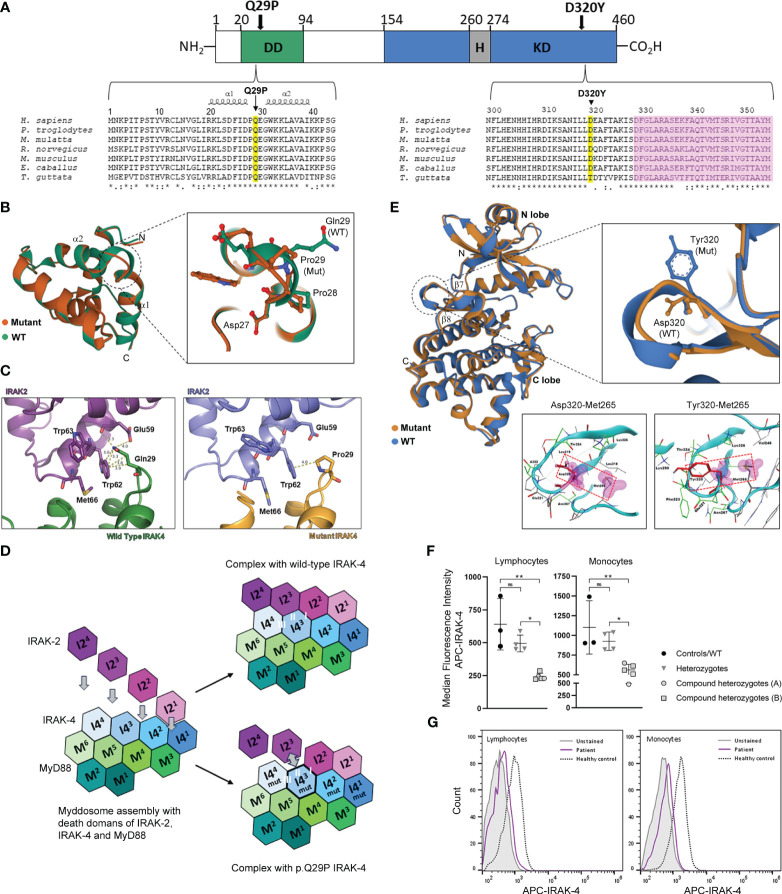
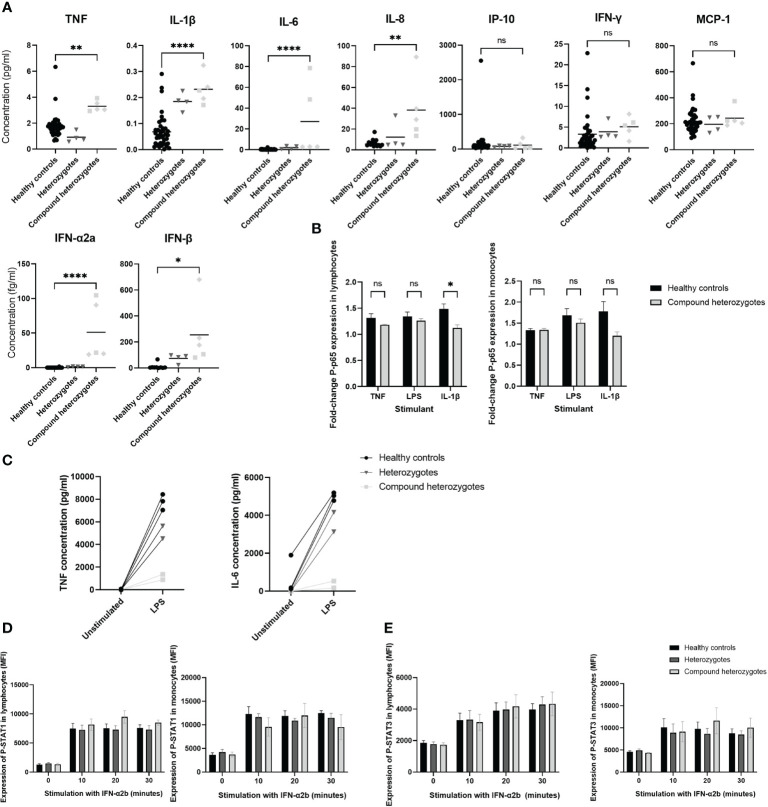
We describe a novel, severe autoinflammatory syndrome characterized by neuroinflammation, systemic autoinflammation, splenomegaly, and anemia (NASA) caused by bi-allelic mutations in IRAK4. IRAK-4 is a serine/threonine kinase with a pivotal role in innate immune signaling from toll-like receptors and production of pro-inflammatory cytokines. In humans, bi-allelic mutations in IRAK4 result in IRAK-4 deficiency and increased susceptibility to pyogenic bacterial infections, but autoinflammation has never been described. We describe 5 affected patients from 2 unrelated families with compound heterozygous mutations in IRAK4 (c.C877T (p.Q293*)/c.G958T (p.D320Y); and c.A86C (p.Q29P)/c.161 + 1G>A) resulting in severe systemic autoinflammation, massive splenomegaly and severe transfusion dependent anemia and, in 3/5 cases, severe neuroinflammation and seizures. IRAK-4 protein expression was reduced in peripheral blood mononuclear cells (PBMC) in affected patients. Immunological analysis demonstrated elevated serum tumor necrosis factor (TNF), interleukin (IL) 1 beta (IL-1β), IL-6, IL-8, interferon α2a (IFN-α2a), and interferon β (IFN-β); and elevated cerebrospinal fluid (CSF) IL-6 without elevation of CSF IFN-α despite perturbed interferon gene signature. Mutations were located within the death domain (DD; p.Q29P and splice site mutation c.161 + 1G>A) and kinase domain (p.Q293*/p.D320Y) of IRAK-4. Structure-based modeling of the DD mutation p.Q29P showed alteration in the alignment of a loop within the DD with loss of contact distance and hydrogen bond interactions with IRAK-1/2 within the myddosome complex. The kinase domain mutation p.D320Y was predicted to stabilize interactions within the kinase active site. While precise mechanisms of autoinflammation in NASA remain uncertain, we speculate that loss of negative regulation of IRAK-4 and IRAK-1; dysregulation of myddosome assembly and disassembly; or kinase active site instability may drive dysregulated IL-6 and TNF production. Blockade of IL-6 resulted in immediate and complete amelioration of systemic autoinflammation and anemia in all 5 patients treated; however, neuroinflammation has, so far proven recalcitrant to IL-6 blockade and the janus kinase (JAK) inhibitor baricitinib, likely due to lack of central nervous system penetration of both drugs. We therefore highlight that bi-allelic mutation in IRAK4 may be associated with a severe and complex autoinflammatory and neuroinflammatory phenotype that we have called NASA (neuroinflammation, autoinflammation, splenomegaly and anemia), in addition to immunodeficiency in humans.
Keywords: IRAK-4; NASA; anemia; autoinflammation; neuroinflammation; splenomegaly; toll-like receptor.
Copyright © 2023 Cooray, Price-Kuehne, Hong, Omoyinmi, Burleigh, Gilmour, Ahmad, Choi, Bahar, Torpiano, Gagunashvili, Jensen, Bellos, Sancho-Shimizu, Herberg, Mankad, Kumar, Kaliakatsos, Worth, Eleftheriou, Whittaker and Brogan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases