

A cross-sectional study on the nasopharyngeal microbiota of individuals with SARS-CoV-2 infection across three COVID-19 waves in India
- PMID: 37744900
- PMCID: PMC10511876
- DOI: 10.3389/fmicb.2023.1238829
A cross-sectional study on the nasopharyngeal microbiota of individuals with SARS-CoV-2 infection across three COVID-19 waves in India
Abstract
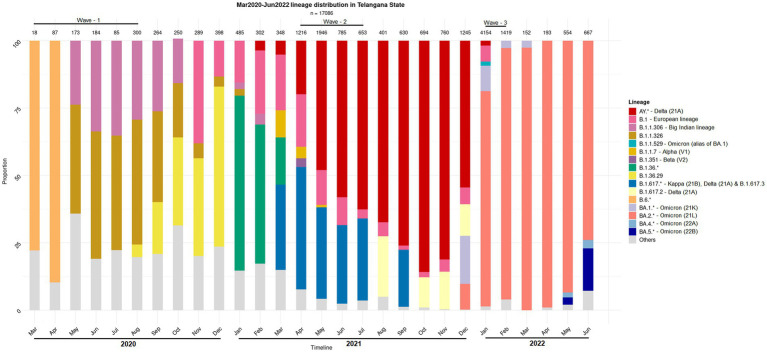
Background: Multiple variants of the SARS-CoV-2 virus have plagued the world through successive waves of infection over the past three years. Independent research groups across geographies have shown that the microbiome composition in COVID-19 positive patients (CP) differs from that of COVID-19 negative individuals (CN). However, these observations were based on limited-sized sample-sets collected primarily from the early days of the pandemic. Here, we study the nasopharyngeal microbiota in COVID-19 patients, wherein the samples have been collected across the three COVID-19 waves witnessed in India, which were driven by different variants of concern.
Methods: The nasopharyngeal swabs were collected from 589 subjects providing samples for diagnostics purposes at the Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, India and subjected to 16s rRNA gene amplicon - based sequencing.
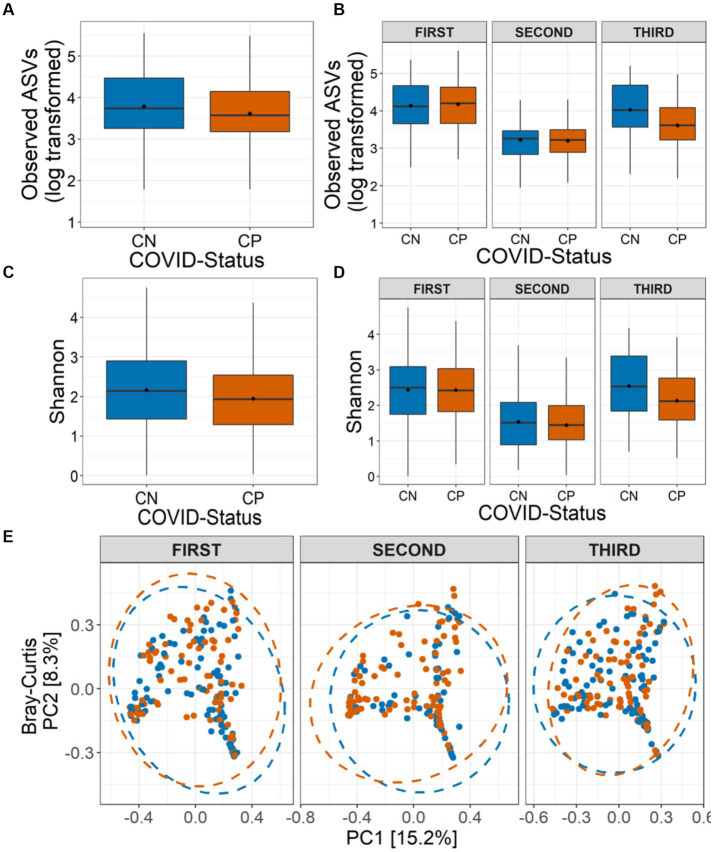
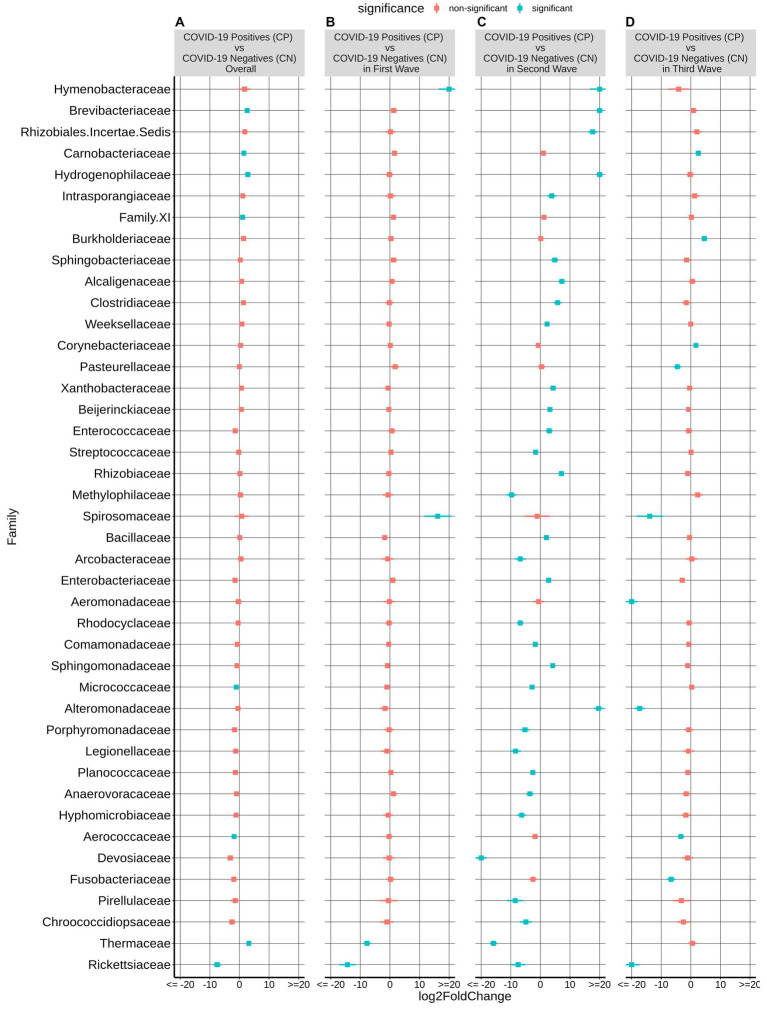
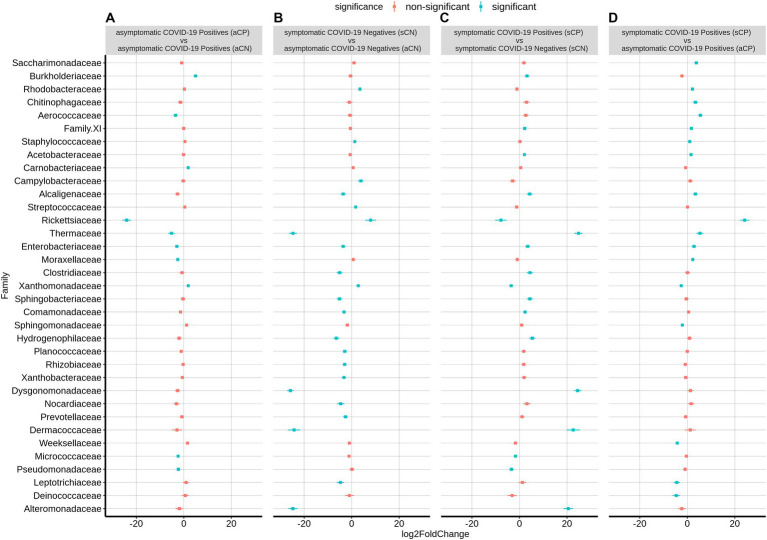
Findings: We found variations in the microbiota of symptomatic vs. asymptomatic COVID-19 patients. CP showed a marked shift in the microbial diversity and composition compared to CN, in a wave-dependent manner. Rickettsiaceae was the only family that was noted to be consistently depleted in CP samples across the waves. The genera Staphylococcus, Anhydrobacter, Thermus, and Aerococcus were observed to be highly abundant in the symptomatic CP patients when compared to the asymptomatic group. In general, we observed a decrease in the burden of opportunistic pathogens in the host microbiota during the later waves of infection.
Interpretation: To our knowledge, this is the first analytical cross-sectional study of this scale, which was designed to understand the relation between the evolving nature of the virus and the changes in the human nasopharyngeal microbiota. Although no clear signatures were observed, this study shall pave the way for a better understanding of the disease pathophysiology and help gather preliminary evidence on whether interventions to the host microbiota can help in better protection or faster recovery.
Keywords: 16S rRNA gene amplicon-based sequencing; COVID-19 disease; Indian cohort; SARS-CoV-2; nasopharyngeal microbiome; variants of concern.
Copyright © 2023 Bose, Wasimuddin, Acharya, Pinna, Kaur, Ranjan, SaiKrishna, Nagabandi, Varma, Tallapaka, Sowpati, Haque, Dutta, Siva and Mande.
Conflict of interest statement
TB, NKP, HK, BV, MMH, AD, and SSM were employed by Tata Consultancy Services Limited. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Nasopharyngeal microbiome reveals the prevalence of opportunistic pathogens in SARS-CoV-2 infected individuals and their association with host types.Microbes Infect. 2022 Feb;24(1):104880. doi: 10.1016/j.micinf.2021.104880. Epub 2021 Aug 21. Microbes Infect. 2022. PMID: 34425246 Free PMC article.
-
Nasopharyngeal microbiota profiling of pregnant women with SARS-CoV-2 infection.Sci Rep. 2022 Aug 4;12(1):13404. doi: 10.1038/s41598-022-17542-z. Sci Rep. 2022. PMID: 35927569 Free PMC article.
-
Alterations in the nasopharyngeal microbiome associated with SARS-CoV-2 infection status and disease severity.PLoS One. 2022 Oct 14;17(10):e0275815. doi: 10.1371/journal.pone.0275815. eCollection 2022. PLoS One. 2022. PMID: 36240246 Free PMC article.
-
The salivary and nasopharyngeal microbiomes are associated with SARS-CoV-2 infection and disease severity.J Med Virol. 2023 Feb;95(2):e28445. doi: 10.1002/jmv.28445. J Med Virol. 2023. PMID: 36583481 Free PMC article.
-
Genomic evolution of the SARS-CoV-2 Variants of Concern: COVID-19 pandemic waves in India.EXCLI J. 2023 Jun 1;22:451-465. doi: 10.17179/excli2023-6098. eCollection 2023. EXCLI J. 2023. PMID: 37534220 Free PMC article. Review.
Cited by
-
The relationship between gut and nasopharyngeal microbiome composition can predict the severity of COVID-19.Elife. 2025 Feb 18;13:RP95292. doi: 10.7554/eLife.95292. Elife. 2025. PMID: 39963971 Free PMC article.
-
Wise Roles and Future Visionary Endeavors of Current Emperor: Advancing Dynamic Methods for Longitudinal Microbiome Meta-Omics Data in Personalized and Precision Medicine.Adv Sci (Weinh). 2024 Dec;11(47):e2400458. doi: 10.1002/advs.202400458. Epub 2024 Nov 13. Adv Sci (Weinh). 2024. PMID: 39535493 Free PMC article. Review.
-
Meta-transcriptomics Reveals Dysbiosis of the Respiratory Microbiome in Older Adults with Long COVID.Research (Wash D C). 2025 Jun 2;8:0720. doi: 10.34133/research.0720. eCollection 2025. Research (Wash D C). 2025. PMID: 40458610 Free PMC article.
-
Nasopharyngeal microbiome composition by SARS-CoV-2 presence and severity.Sci Rep. 2025 Jul 2;15(1):23185. doi: 10.1038/s41598-025-01764-y. Sci Rep. 2025. PMID: 40603926 Free PMC article.
-
Alterations in microbiota of patients with COVID-19: implications for therapeutic interventions.MedComm (2020). 2024 Mar 15;5(4):e513. doi: 10.1002/mco2.513. eCollection 2024 Apr. MedComm (2020). 2024. PMID: 38495122 Free PMC article. Review.
References
-
- Alauzet C., Aujoulat F., Lozniewski A., Ben Brahim S., Domenjod C., Enault C., et al. (2021). A new look at the genus Solobacterium: a retrospective analysis of twenty-seven cases of infection involving S. moorei and a Review of Sequence Databases and the Literature. Microorganisms 9:1229. doi: 10.3390/microorganisms9061229, PMID: - DOI - PMC - PubMed
-
- Aleem A., Akbar Samad A. B., Slenker A. K. (2022). Emerging variants of SARS-CoV-2 and novel therapeutics against coronavirus (COVID-19). Stat Pearls. Treasure Island, FL. - PubMed
-
- Barre A., Van Damme E. J. M., Simplicien M., Le Poder S., Klonjkowski B., Benoist H., et al. (2021). Man-specific lectins from plants, Fungi, algae and Cyanobacteria, as potential blockers for SARS-CoV, MERS-CoV and SARS-CoV-2 (COVID-19) coronaviruses: biomedical perspectives. Cells 10:1619. doi: 10.3390/cells10071619, PMID: - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous