

An integrated transcriptomic and metabolic phenotype analysis to uncover the metabolic characteristics of a genetically engineered Candida utilis strain expressing δ-zein gene
- PMID: 37744922
- PMCID: PMC10513430
- DOI: 10.3389/fmicb.2023.1241462
An integrated transcriptomic and metabolic phenotype analysis to uncover the metabolic characteristics of a genetically engineered Candida utilis strain expressing δ-zein gene
Abstract
Introduction: Candida utilis (C. utilis) has been extensively utilized as human food or animal feed additives. With its ability to support heterologous gene expression, C. utilis proves to be a valuable platform for the synthesis of proteins and metabolites that possess both high nutritional and economic value. However, there remains a dearth of research focused on the characteristics of C. utilis through genomic, transcriptomic and metabolic approaches.
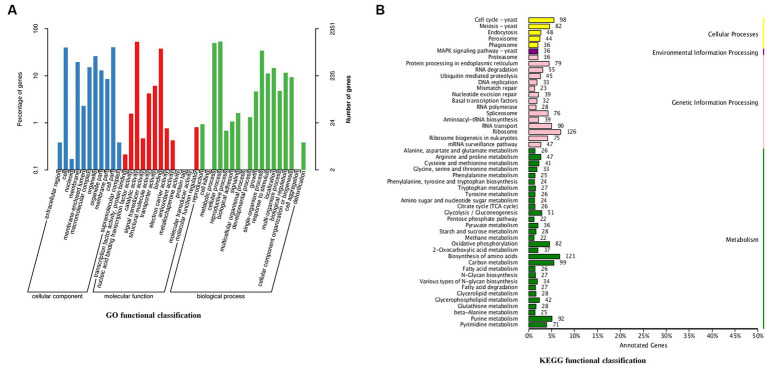
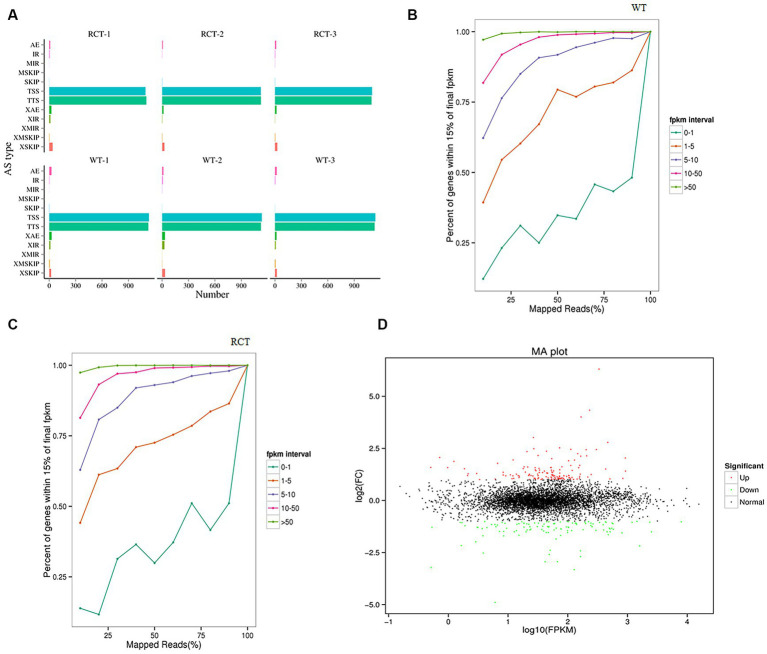
Methods: With the aim of unraveling the molecular mechanism and genetic basis governing the biological process of C. utilis, we embarked on a de novo sequencing endeavor to acquire comprehensive sequence data. In addition, an integrated transcriptomic and metabolic phenotype analysis was performed to compare the wild-type C. utilis (WT) with a genetically engineered strain of C. utilis that harbors the heterologous δ-zein gene (RCT).
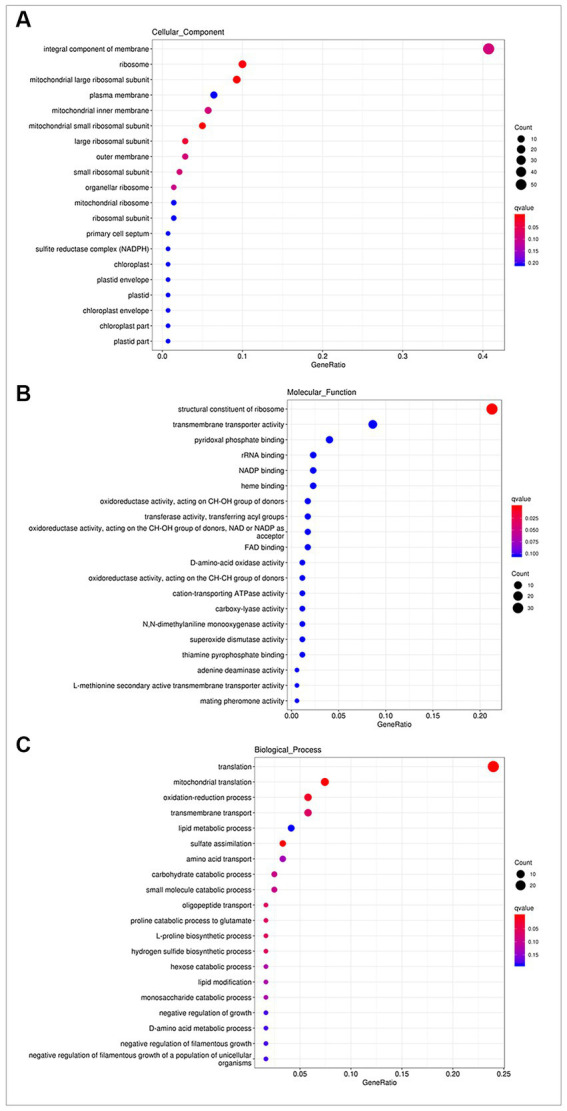
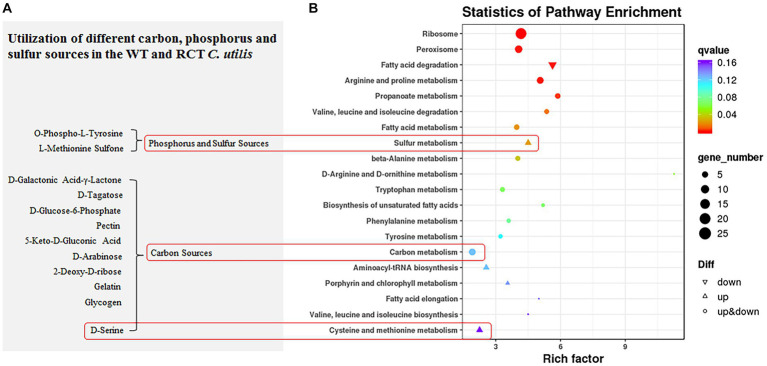
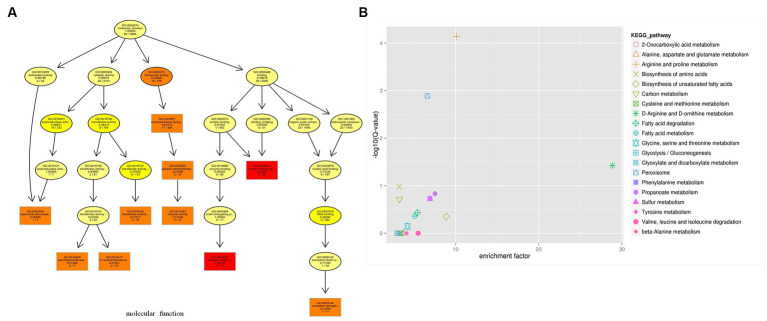
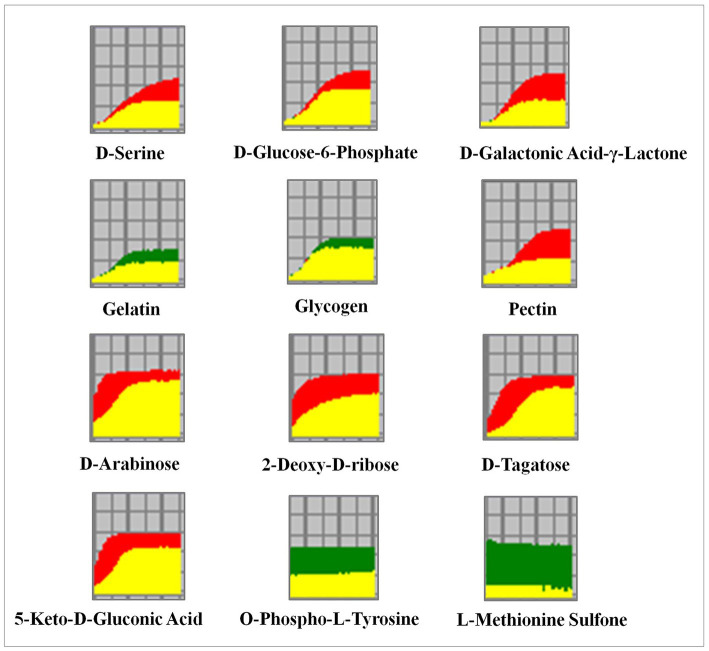
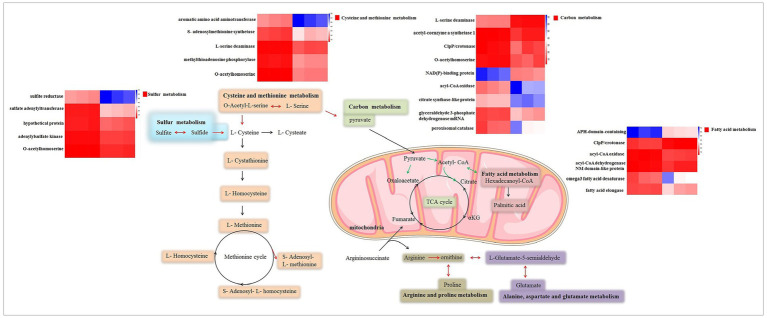
Results: δ-zein is a protein rich in methionine found in the endosperm of maize. The integrated analysis of transcriptomic and metabolic phenotypes uncovered significant metabolic diversity between the WT and RCT C. utilis. A total of 252 differentially expressed genes were identified, primarily associated with ribosome function, peroxisome activity, arginine and proline metabolism, carbon metabolism, and fatty acid degradation. In the experimental setup using PM1, PM2, and PM4 plates, a total of 284 growth conditions were tested. A comparison between the WT and RCT C. utilis demonstrated significant increases in the utilization of certain carbon source substrates by RCT. Gelatin and glycogen were found to be significantly utilized to a greater extent by RCT compared to WT. Additionally, in terms of sulfur source substrates, RCT exhibited significantly increased utilization of O-Phospho-L-Tyrosine and L-Methionine Sulfone when compared to WT.
Discussion: The introduction of δ-zein gene into C. utilis may lead to significant changes in the metabolic substrates and metabolic pathways, but does not weaken the activity of the strain. Our study provides new insights into the transcriptomic and metabolic characteristics of the genetically engineered C. utilis strain harboring δ-zein gene, which has the potential to advance the utilization of C. utilis as an efficient protein feed in agricultural applications.
Keywords: Candida utilis; genome; metabolic phenotype; transcriptome; δ-zein.
Copyright © 2023 He, Gong, Wan, Hu and Yu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
An optimised promoter and signal peptide improves methionine production of a genetically engineered Candida utilis harboring the δ-zein gene.Front Microbiol. 2025 Jul 4;16:1586229. doi: 10.3389/fmicb.2025.1586229. eCollection 2025. Front Microbiol. 2025. PMID: 40687855 Free PMC article.
-
Expression of an 11 kDa methionine-rich delta-zein in transgenic soybean results in the formation of two types of novel protein bodies in transitional cells situated between the vascular tissue and storage parenchyma cells.Plant Biotechnol J. 2004 May;2(3):199-210. doi: 10.1111/j.1467-7652.2004.00063.x. Plant Biotechnol J. 2004. PMID: 17147611
-
Metabolomic and transcriptomic analysis for rate-limiting metabolic steps in xylose utilization by recombinant Candida utilis.Biosci Biotechnol Biochem. 2013;77(7):1441-8. doi: 10.1271/bbb.130093. Epub 2013 Jul 7. Biosci Biotechnol Biochem. 2013. PMID: 23832335
-
Candida utilis and Cyberlindnera (Pichia) jadinii: yeast relatives with expanding applications.Appl Microbiol Biotechnol. 2016 Aug;100(16):6981-90. doi: 10.1007/s00253-016-7700-8. Epub 2016 Jun 29. Appl Microbiol Biotechnol. 2016. PMID: 27357226 Review.
-
Safety and nutritional assessment of GM plants and derived food and feed: the role of animal feeding trials.Food Chem Toxicol. 2008 Mar;46 Suppl 1:S2-70. doi: 10.1016/j.fct.2008.02.008. Epub 2008 Feb 13. Food Chem Toxicol. 2008. PMID: 18328408 Review.
Cited by
-
Transcriptomic and metabolomic correlation analysis: effect of initial SO2 addition on higher alcohol synthesis in Saccharomyces cerevisiae and identification of key regulatory genes.Front Microbiol. 2024 May 13;15:1394880. doi: 10.3389/fmicb.2024.1394880. eCollection 2024. Front Microbiol. 2024. PMID: 38803372 Free PMC article.
References
-
- Acosta D. A. V., Denicol A. C., Tribulo P., Rivelli M. I., Skenandore C., Zhou Z., et al. . (2016). Effects of rumen-protected methionine and choline supplementation on the preimplantation embryo in Holstein cows. Theriogenology 85, 1669–1679. doi: 10.1016/j.theriogenology.2016.01.024, PMID: - DOI - PubMed
-
- Bagga S., Armendaris A., Klypina N., Ray I., Ghoshroy S., Endress M., et al. . (2004). Genetic engineering ruminal stable high methionine protein in the foliage of alfalfa. Int. J. Plant Sci. 166, 273–283. doi: 10.1016/j.plantsci.2003.09.014 - DOI
LinkOut - more resources
Full Text Sources