

Investigation of the individual genetic evolution of SARS-CoV-2 in a small cluster during the rapid spread of the BF.5 lineage in Tokyo, Japan
- PMID: 37744926
- PMCID: PMC10516552
- DOI: 10.3389/fmicb.2023.1229234
Investigation of the individual genetic evolution of SARS-CoV-2 in a small cluster during the rapid spread of the BF.5 lineage in Tokyo, Japan
Abstract
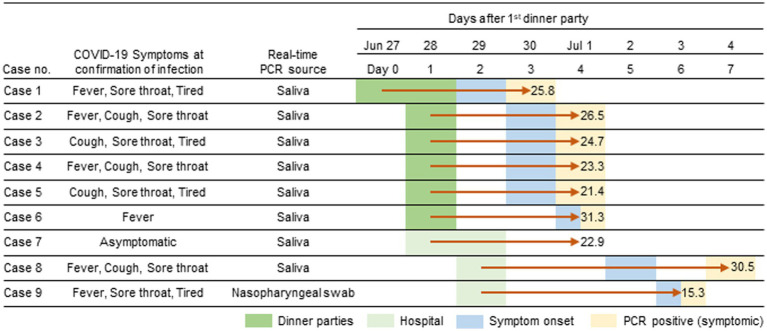
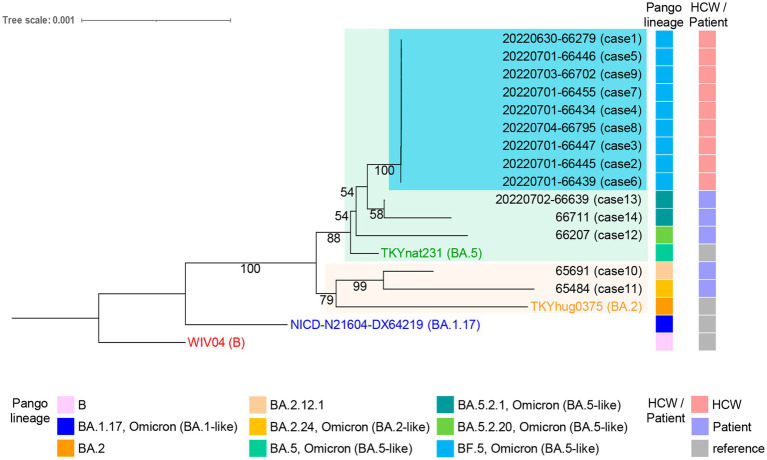
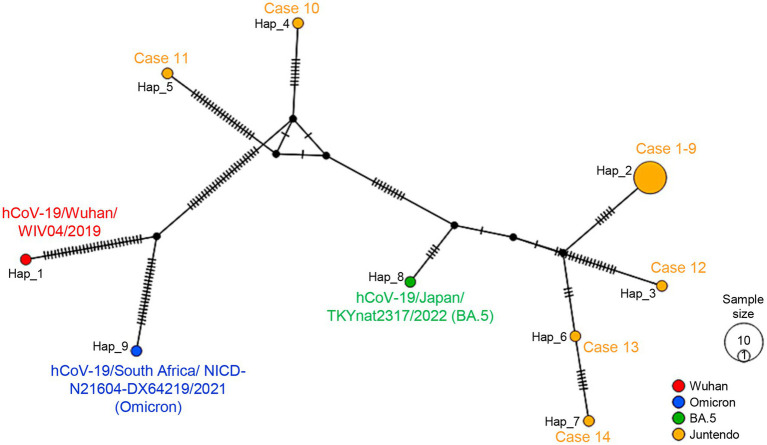
There has been a decreasing trend in new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) cases and fatalities worldwide. The virus has been evolving, indicating the potential emergence of new variants and uncertainties. These challenges necessitate continued efforts in disease control and mitigation strategies. We investigated a small cluster of SARS-CoV-2 Omicron variant infections containing a common set of genomic mutations, which provided a valuable model for investigating the transmission mechanism of genetic alterations. We conducted a study at a medical center in Japan during the Omicron surge (sub-lineage BA.5), sequencing the entire SARS-CoV-2 genomes from infected individuals and evaluating the phylogenetic tree and haplotype network among the variants. We compared the mutations present in each strain within the BA.5 strain, TKYnat2317, which was first identified in Tokyo, Japan. From June 29th to July 4th 2022, nine healthcare workers (HCWs) tested positive for SARS-CoV-2 by real-time PCR. During the same period, five patients also tested positive by real-time PCR. Whole genome sequencing revealed that the infected patients belonged to either the isolated BA.2 or BA.5 sub-lineage, while the healthcare worker infections were classified as BF.5. The phylogenetic tree and haplotype network clearly showed the specificity and similarity of the HCW cluster. We identified 12 common mutations in the cluster, including I110V in nonstructural protein 4 (nsp4), A1020S in the Spike protein, and H47Y in ORF7a, compared to the BA.5 reference. Additionally, one case had the extra nucleotide-deletion mutation I27* in ORF10, and low frequencies of genetic alterations were also found in certain instances. The results of genome sequencing showed that the nine HCWs shared a set of genetic mutations, indicating transmission within the cluster. Minor mutations observed in five HCW individuals suggested the emergence of new virus variants. Five amino acid substitutions occurred in nsp3, which could potentially affect virus replication or immune escape. Intra-host evolution also generated additional mutations. The cluster exhibited a mild disease course, with individuals in this case, recovering without requiring any medical treatments. Further investigation is needed to understand the relationship between the genetic evolution of the virus and the symptoms.
Keywords: BF.5 lineage; SARS-CoV-2; cluster; gene mutation; intra-host evolution; omicron variant; whole genome sequencing; within-host diversity.
Copyright © 2023 Jin, Oyama, Tabe, Tsuchiya, Hando, Wakita, Yan, Saita, Takei, Horiuchi, Miida, Naito, Takahashi and Ogawa.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Ahmadi K., Shahbazi B., Zakeri A. J., Gouklani H. (2022). Characterization of SARS-CoV-2 omicron variants from Iran and evaluation of the effect of mutations on the spike, nucleocapsid, ORF8, and ORF9b proteins function. J. Biomol. Struct. Dyn. 1-16, 1–16. doi: 10.1080/07391102.2022.2162131, PMID: - DOI - PubMed
-
- Chatterjee S., Bhattacharya M., Nag S., Dhama K., Chakraborty C. (2023). A detailed overview of SARS-CoV-2 omicron: its sub-variants, mutations and pathophysiology, clinical characteristics, immunological landscape, immune escape, and therapies. Viruses 15:167. doi: 10.3390/v15010167, PMID: - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous