

This is a preprint.
bamSliceR: a Bioconductor package for rapid, cross-cohort variant and allelic bias analysis
- PMID: 37745420
- PMCID: PMC10516001
- DOI: 10.1101/2023.09.15.558026
bamSliceR: a Bioconductor package for rapid, cross-cohort variant and allelic bias analysis
Update in
-
bamSliceR: a Bioconductor package for rapid, cross-cohort variant and allelic bias analysis.Bioinform Adv. 2025 Apr 28;5(1):vbaf098. doi: 10.1093/bioadv/vbaf098. eCollection 2025. Bioinform Adv. 2025. PMID: 40395503 Free PMC article.
Abstract
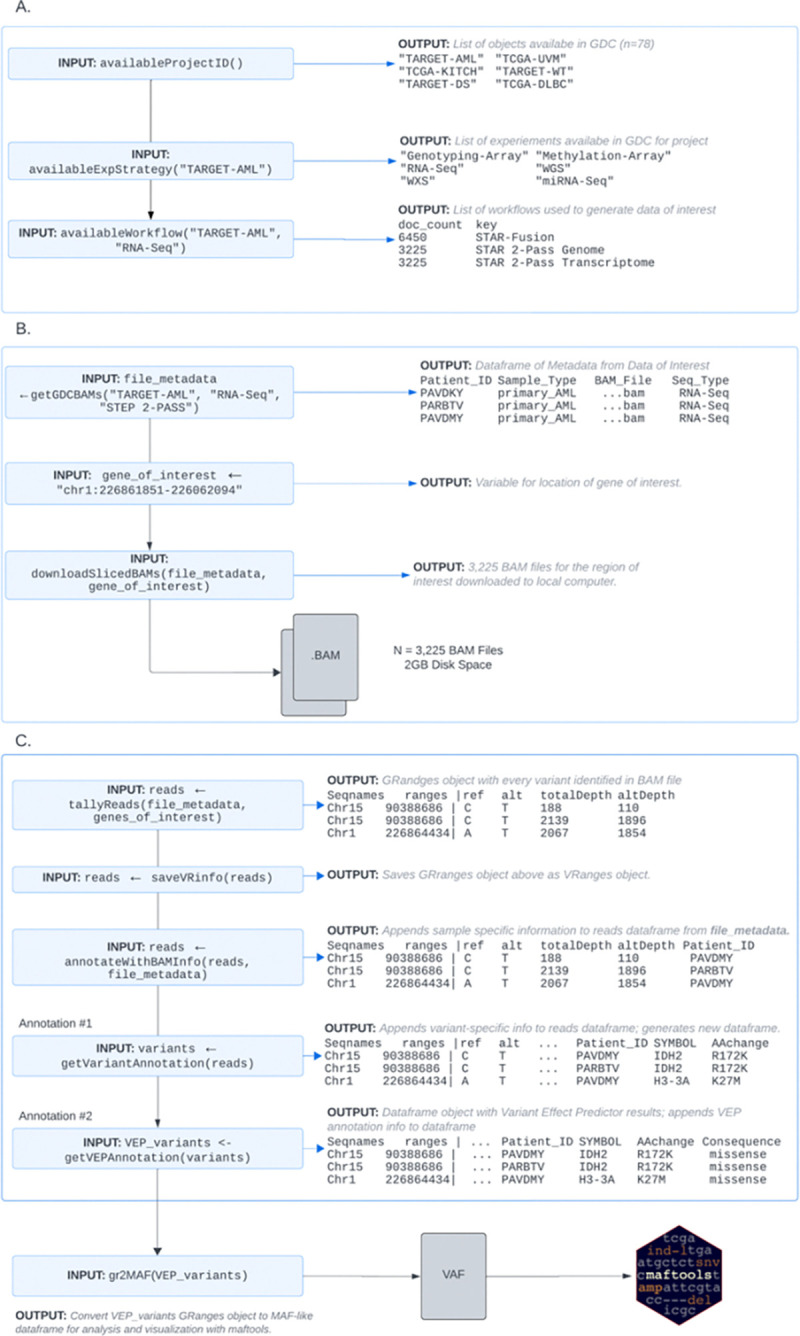
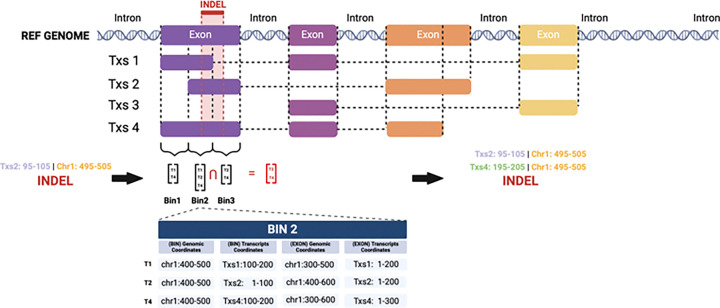
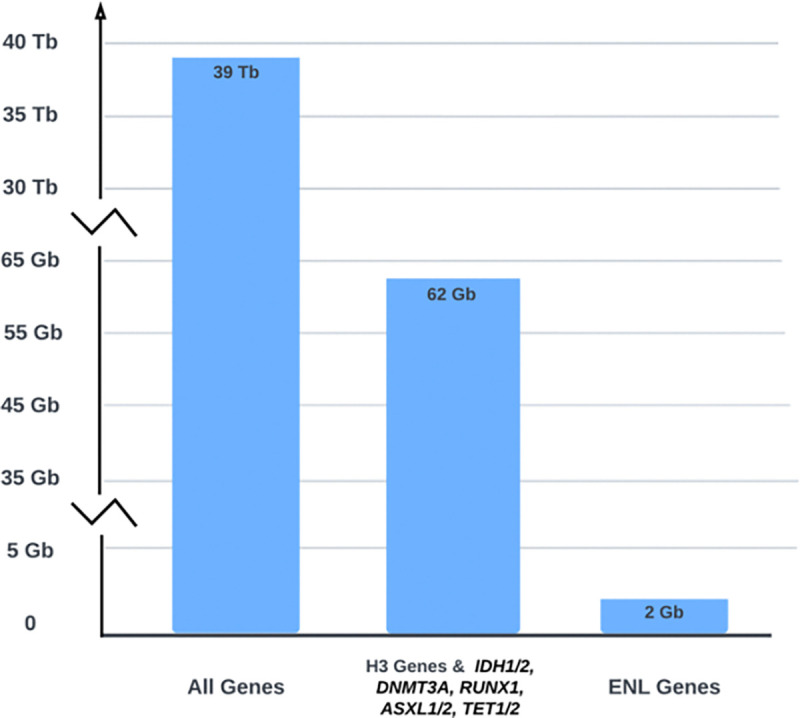
The NCI Genomic Data Commons (GDC) provides controlled access to sequencing data from thousands of subjects, enabling large-scale study of impactful genetic alterations such as simple and complex germline and structural variants. However, efficient analysis requires significant computational resources and expertise, especially when recalling variants from raw sequence reads. We thus developed bamSliceR , an R/Bioconductor package that builds upon the GenomicDataCommons package to extract aligned sequence reads from cross-GDC meta-cohorts, followed by targeted analysis of variants and effects (including transcript-aware variant annotation from transcriptome-aligned GDC RNA data). Here we demonstrate population-scale genomic & transcriptomic analyses with minimal compute burden via bamSliceR , identifying recurrent, clinically relevant sequence and structural variants in the TARGET AML and BEAT-AML cohorts. We then validate results in the (non-GDC) Leucegene cohort, demonstrating how the bamSliceR pipeline can be seamlessly applied to replicate findings in non-GDC cohorts. These variants directly yield clinically impactful and biologically testable hypotheses for mechanistic investigation. bamSliceR has been submitted to the Bioconductor project, where it is presently under review, and is available on GitHub at https://github.com/trichelab/bamSliceR.
Conflict of interest statement
Conflict of interest None declared.
Figures



References
-
- Schwartzentruber J. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources