

This is a preprint.
Single-cell nascent RNA sequencing using click-chemistry unveils coordinated transcription
- PMID: 37745427
- PMCID: PMC10516050
- DOI: 10.1101/2023.09.15.558015
Single-cell nascent RNA sequencing using click-chemistry unveils coordinated transcription
Update in
-
Single-cell nascent RNA sequencing unveils coordinated global transcription.Nature. 2024 Jul;631(8019):216-223. doi: 10.1038/s41586-024-07517-7. Epub 2024 Jun 5. Nature. 2024. PMID: 38839954 Free PMC article.
Abstract
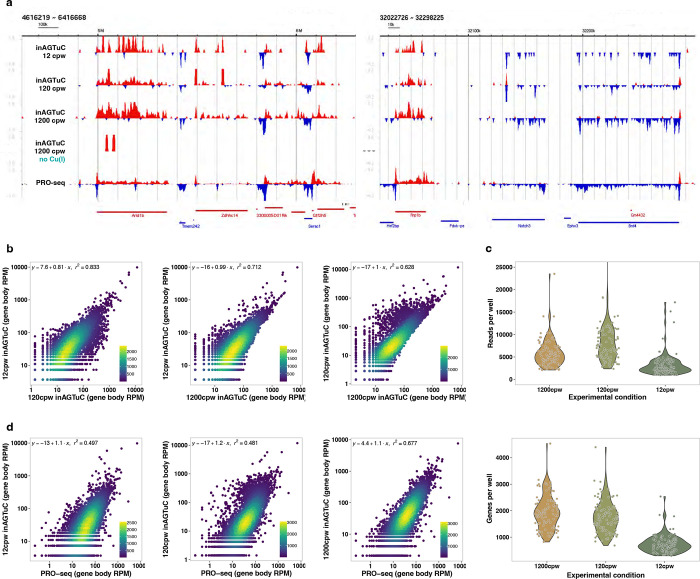
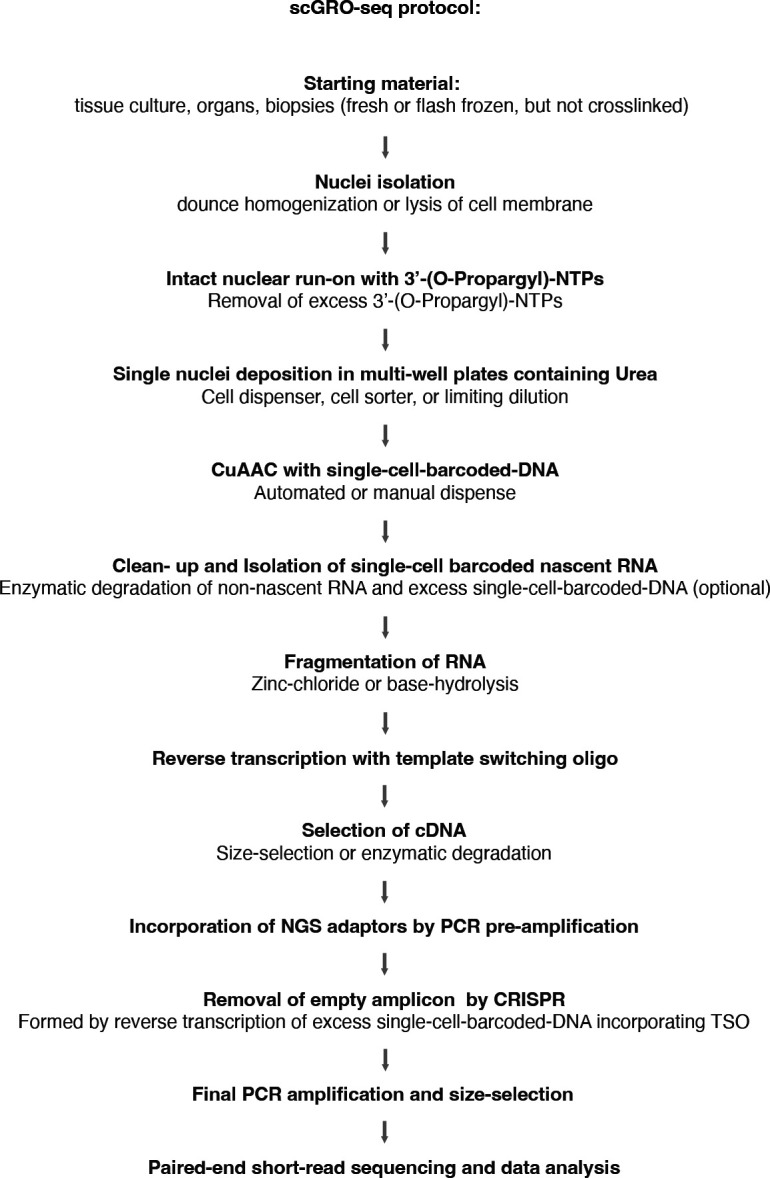
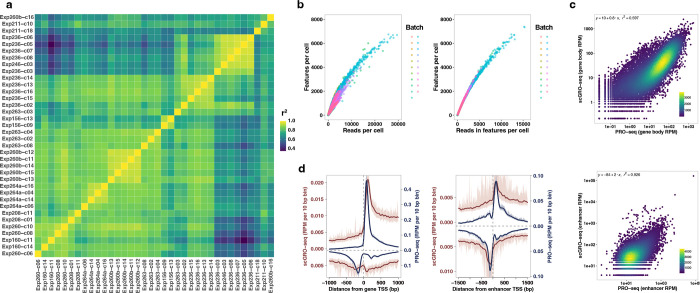
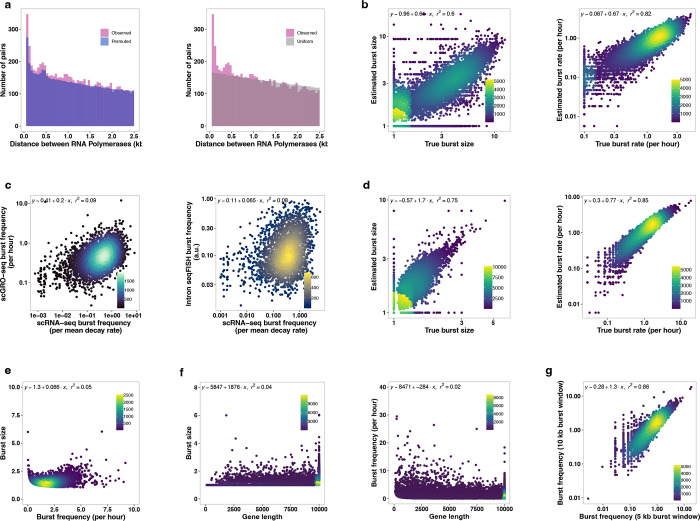
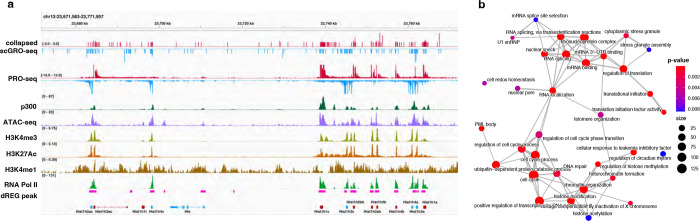
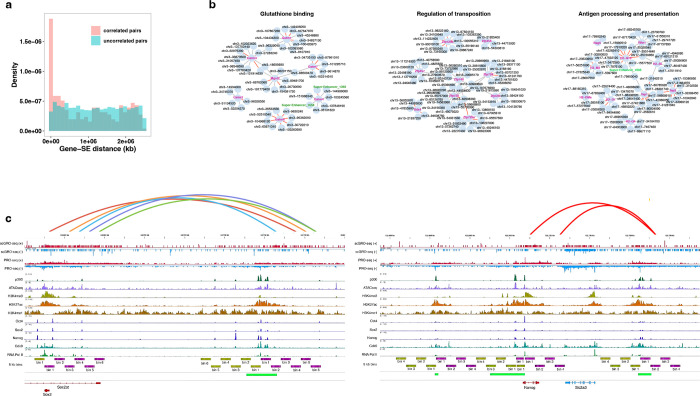
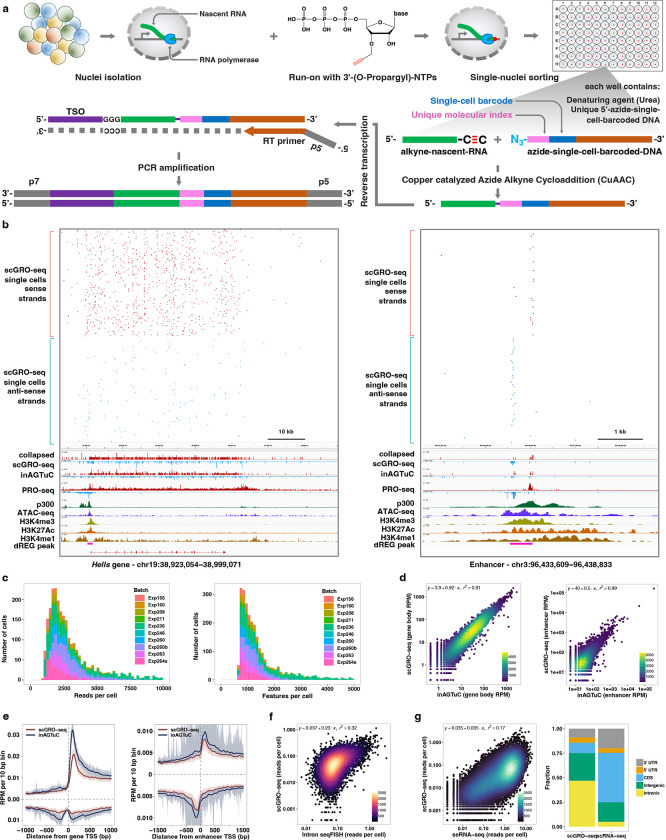
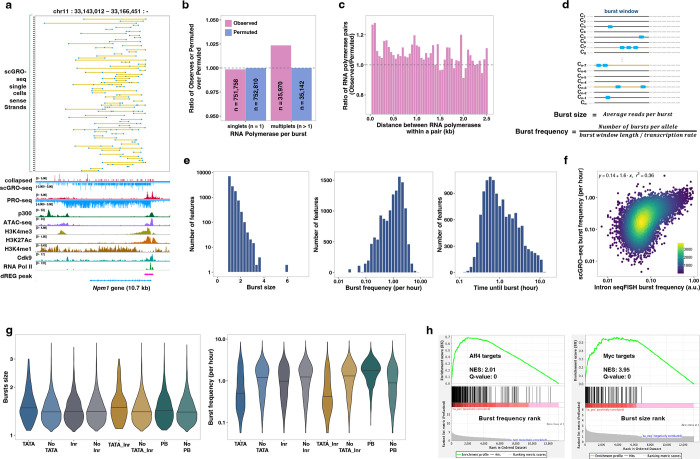
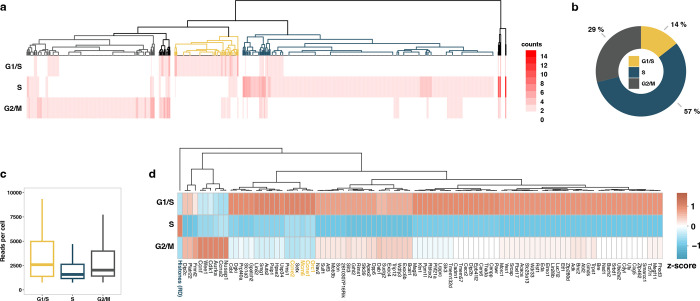
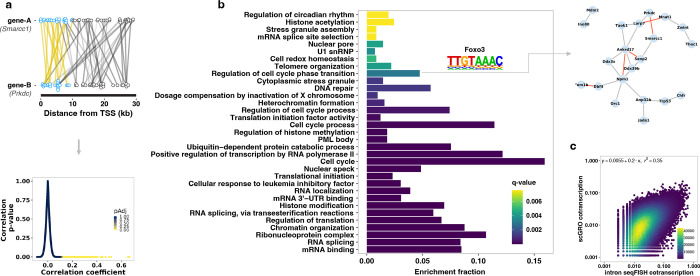
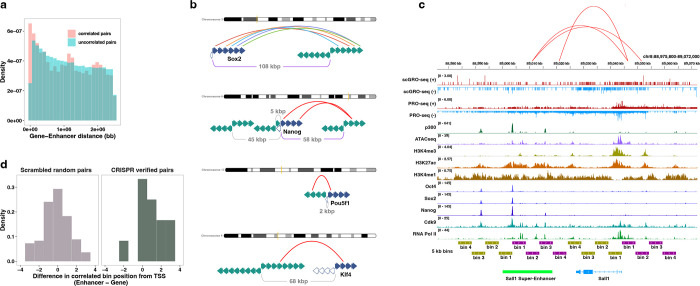
Transcription is the primary regulatory step in gene expression. Divergent transcription initiation from promoters and enhancers produces stable RNAs from genes and unstable RNAs from enhancers1-5. Nascent RNA capture and sequencing assays simultaneously measure gene and enhancer activity in cell populations6-9. However, fundamental questions in the temporal regulation of transcription and enhancer-gene synchrony remain unanswered primarily due to the absence of a single-cell perspective on active transcription. In this study, we present scGRO-seq - a novel single-cell nascent RNA sequencing assay using click-chemistry - and unveil the coordinated transcription throughout the genome. scGRO-seq demonstrates the episodic nature of transcription, and estimates burst size and frequency by directly quantifying transcribing RNA polymerases in individual cells. It reveals the co-transcription of functionally related genes and leverages the replication-dependent non-polyadenylated histone genes transcription to elucidate cell-cycle dynamics. The single-nucleotide spatial and temporal resolution of scGRO-seq identifies networks of enhancers and genes and indicates that the bursting of transcription at super-enhancers precedes the burst from associated genes. By imparting insights into the dynamic nature of transcription and the origin and propagation of transcription signals, scGRO-seq demonstrates its unique ability to investigate the mechanisms of transcription regulation and the role of enhancers in gene expression.
Conflict of interest statement
Competing interests US patent number US-11519027-B2 on ‘SINGLE-CELL RNA SEQUENCING USING CLICK-CHEMISTRY” was granted on December 6, 2022, to the Massachusetts Institute of Technology, Cambridge, MA (US), on which P.A.S. and D.B.M. are inventors. The authors declare no other competing interests.
Figures
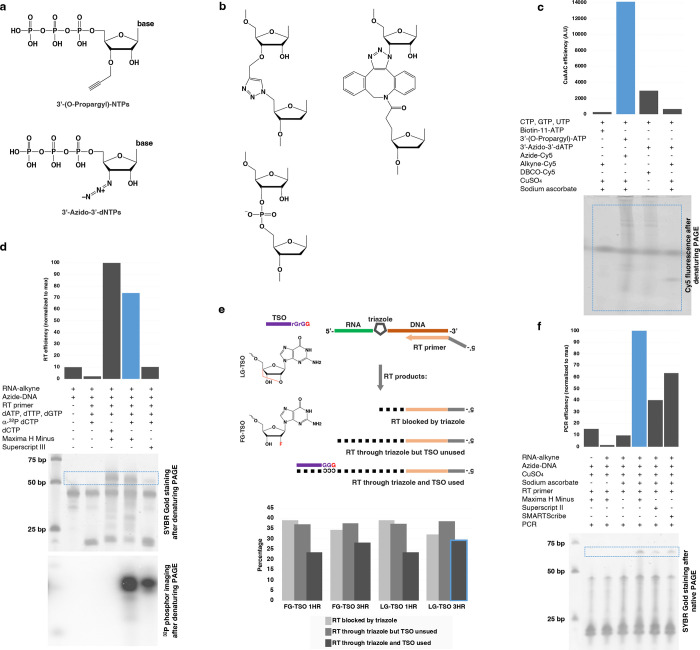
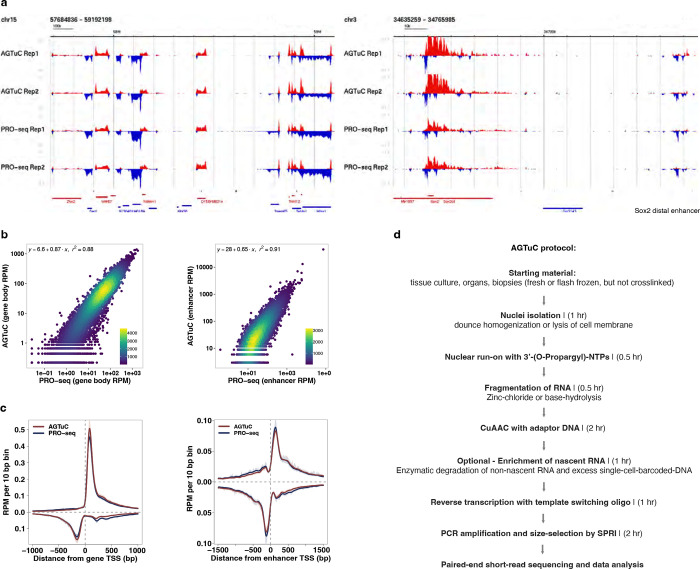
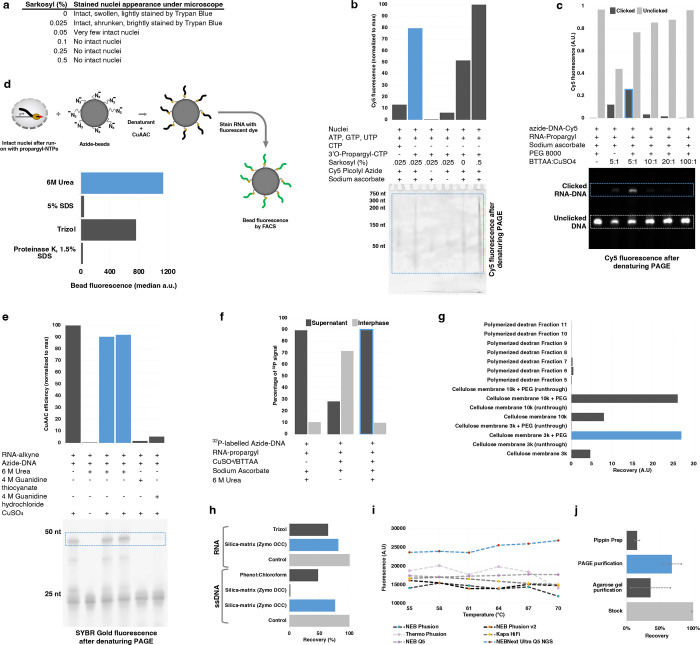
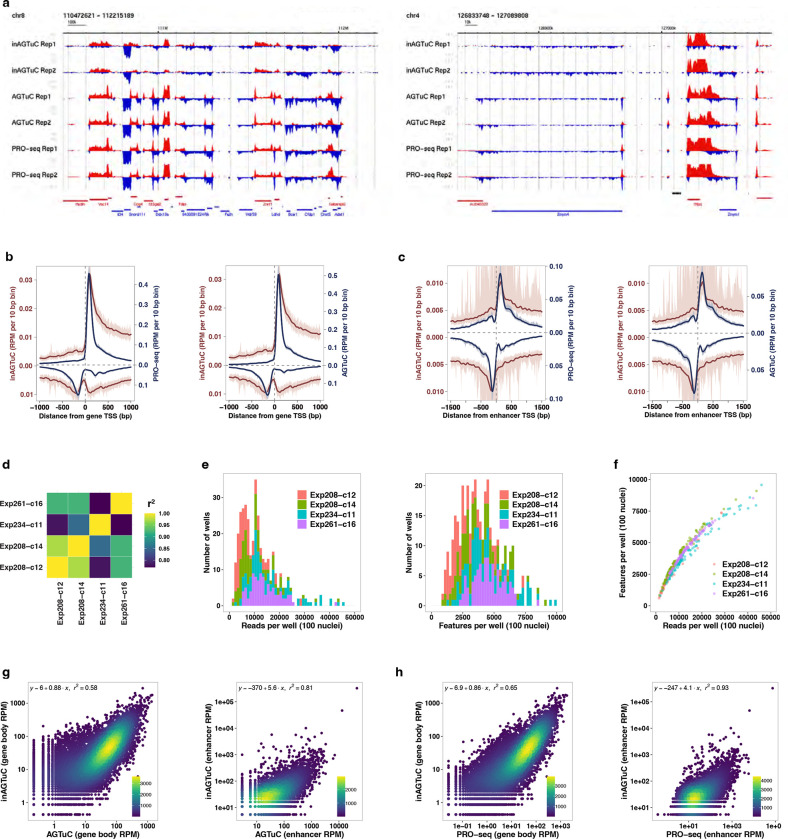
References
-
- Kapranov P., Willingham A. T. & Gingeras T. R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 8, 413–423 (2007). - PubMed
-
- Kapranov P. et al. RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science 316, 1484–1488 (2007). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources