

This is a preprint.
Systematic identification of disease-causing promoter and untranslated region variants in 8,040 undiagnosed individuals with rare disease
- PMID: 37745552
- PMCID: PMC10516070
- DOI: 10.1101/2023.09.12.23295416
Systematic identification of disease-causing promoter and untranslated region variants in 8,040 undiagnosed individuals with rare disease
Update in
-
Systematic identification of disease-causing promoter and untranslated region variants in 8040 undiagnosed individuals with rare disease.Genome Med. 2025 Apr 14;17(1):40. doi: 10.1186/s13073-025-01464-2. Genome Med. 2025. PMID: 40229884 Free PMC article.
Abstract
Background: Both promoters and untranslated regions (UTRs) have critical regulatory roles, yet variants in these regions are largely excluded from clinical genetic testing due to difficulty in interpreting pathogenicity. The extent to which these regions may harbour diagnoses for individuals with rare disease is currently unknown.
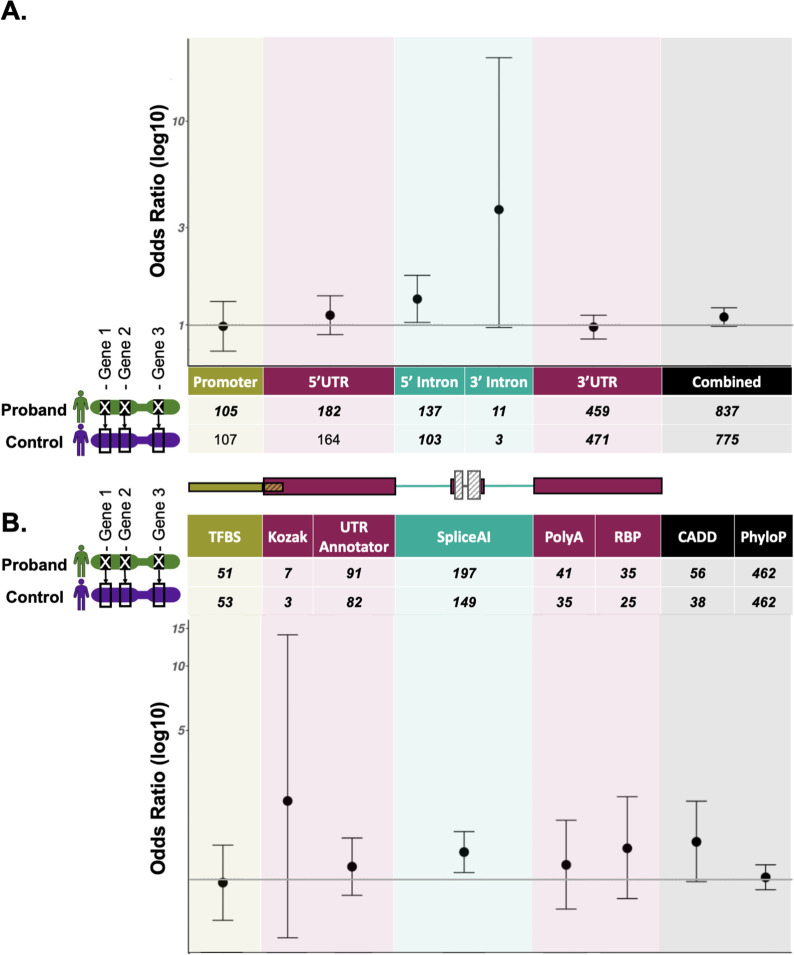
Methods: We present a framework for the identification and annotation of potentially deleterious proximal promoter and UTR variants in known dominant disease genes. We use this framework to annotate de novo variants (DNVs) in 8,040 undiagnosed individuals in the Genomics England 100,000 genomes project, which were subject to strict region-based filtering, clinical review, and validation studies where possible. In addition, we performed region and variant annotation-based burden testing in 7,862 unrelated probands against matched unaffected controls.
Results: We prioritised eleven DNVs and identified an additional variant overlapping one of the eleven. Ten of these twelve variants (82%) are in genes that are a strong match to the individual's phenotype and six had not previously been identified. Through burden testing, we did not observe a significant enrichment of potentially deleterious promoter and/or UTR variants in individuals with rare disease collectively across any of our region or variant annotations.
Conclusions: Overall, we demonstrate the value of screening promoters and UTRs to uncover additional diagnoses for previously undiagnosed individuals with rare disease and provide a framework for doing so without dramatically increasing interpretation burden.
Keywords: Untranslated regions; non-coding; promoters; rare disease; regulatory regions; splicing.
Figures


References
-
- Kircher M, Xiong C, Martin B, Schubach M, Inoue F, Bell RJA, et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat Commun [Internet]. 2019. Aug 8 [cited 2023 Sep 12];10(1). Available from: https://pubmed.ncbi.nlm.nih.gov/31395865/ - PMC - PubMed
-
- Griesemer D, Xue JR, Reilly SK, Ulirsch JC, Kukreja K, Davis JR, et al. Genome-wide functional screen of 3’UTR variants uncovers causal variants for human disease and evolution. Cell [Internet]. 2021. Sep 30 [cited 2023 Sep 12];184(20). Available from: https://pubmed.ncbi.nlm.nih.gov/34534445/ - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources