

This is a preprint.
Pyruvate Kinase Activity Regulates Cystine Starvation Induced Ferroptosis through Malic Enzyme 1 in Pancreatic Cancer Cells
- PMID: 37745559
- PMCID: PMC10516027
- DOI: 10.1101/2023.09.15.557984
Pyruvate Kinase Activity Regulates Cystine Starvation Induced Ferroptosis through Malic Enzyme 1 in Pancreatic Cancer Cells
Abstract
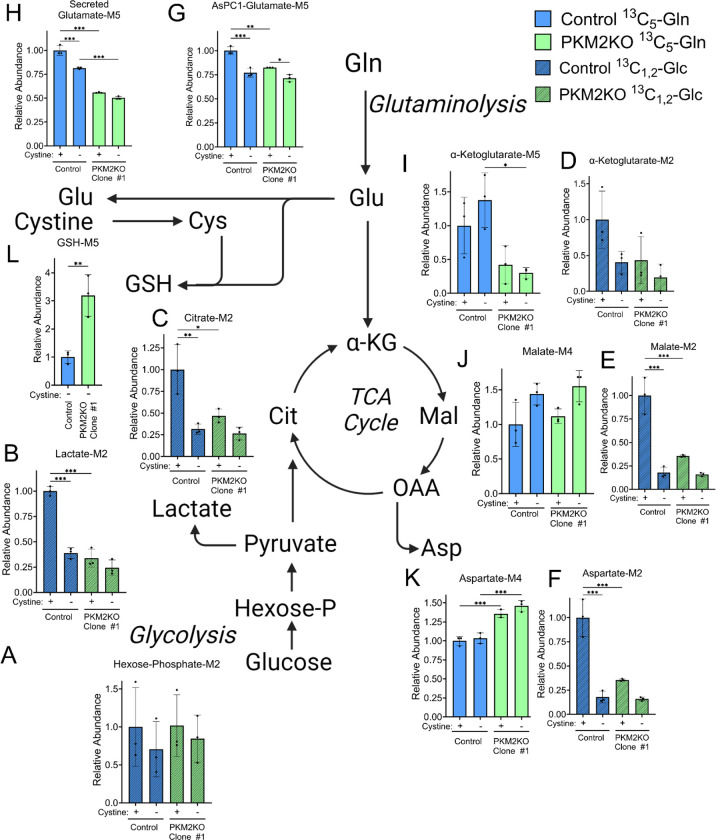
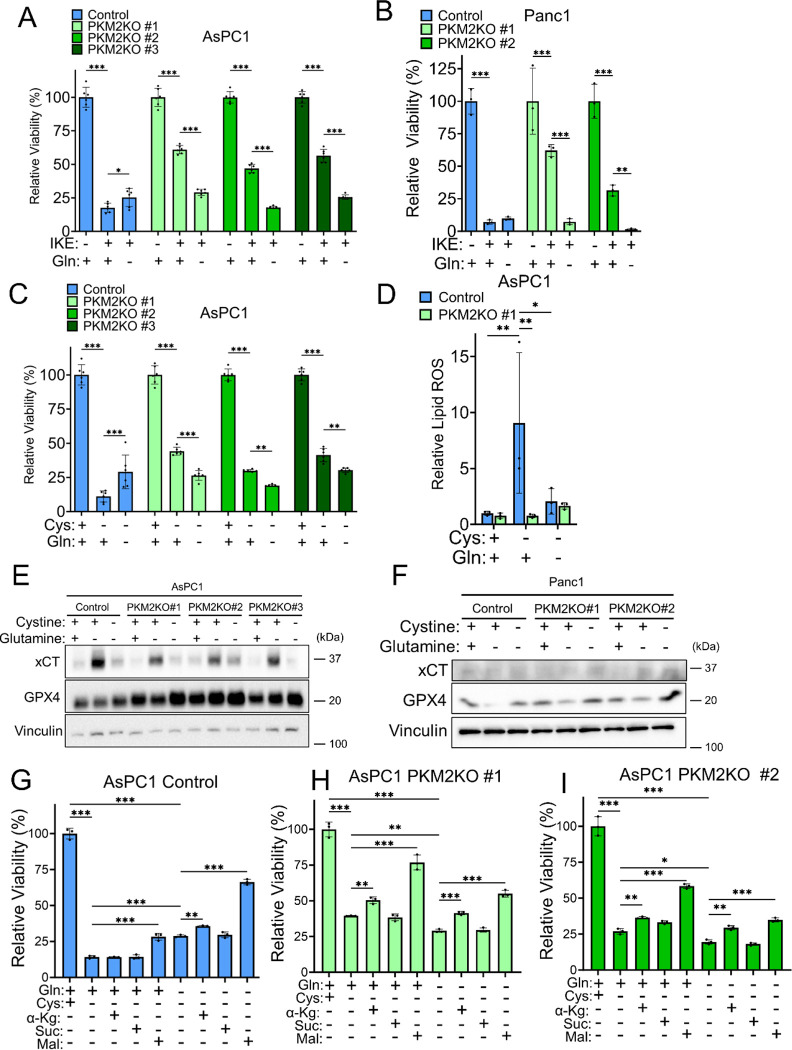
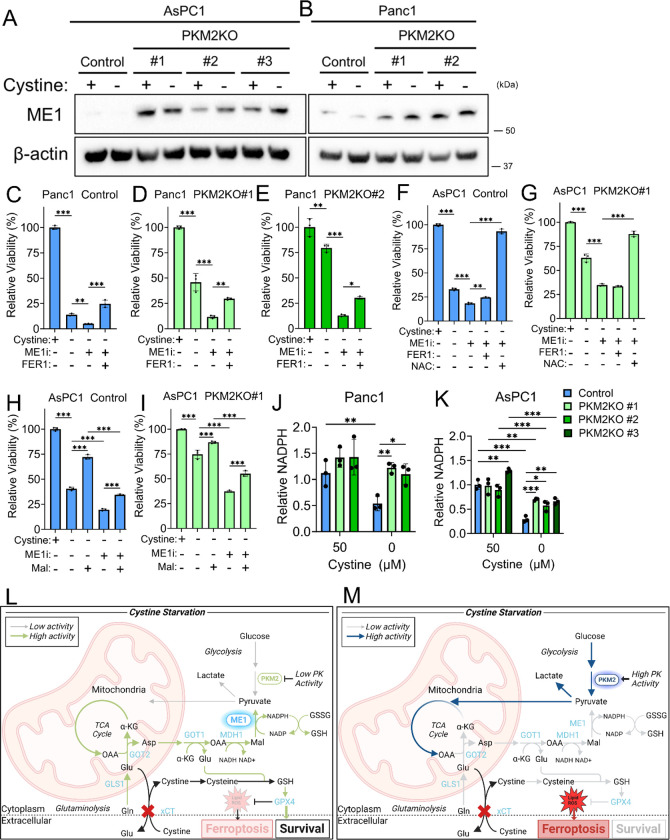
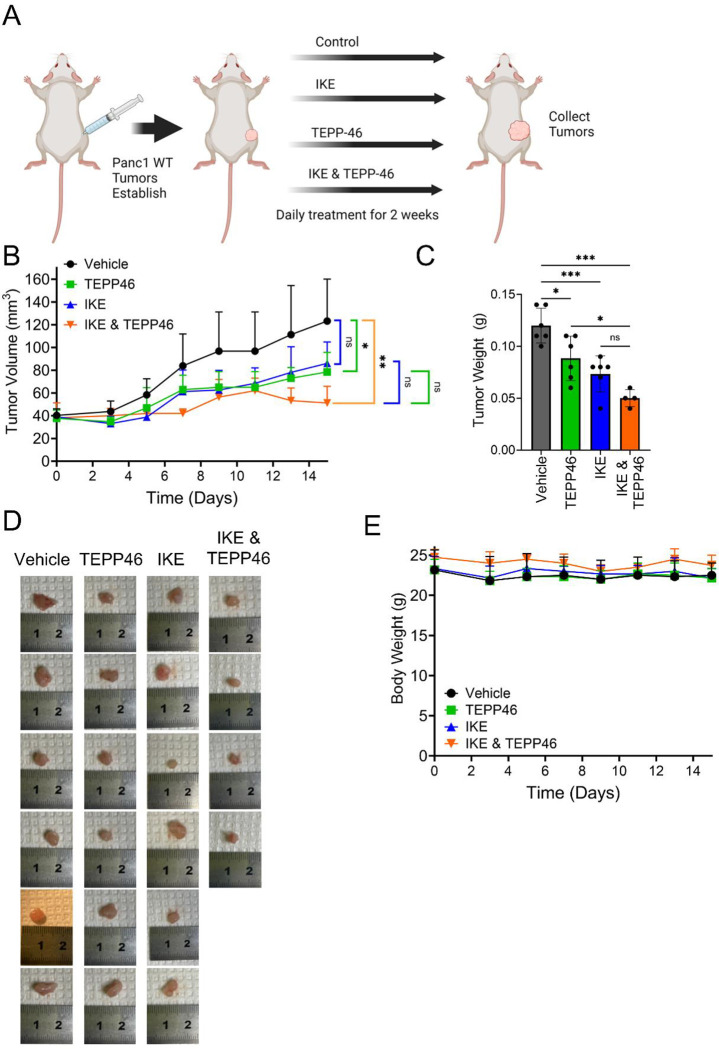
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer with high mortality and limited efficacious therapeutic options. PDAC cells undergo metabolic alterations to survive within a nutrient-depleted tumor microenvironment. One critical metabolic shift in PDAC cells occurs through altered isoform expression of the glycolytic enzyme, pyruvate kinase (PK). Pancreatic cancer cells preferentially upregulate pyruvate kinase muscle isoform 2 isoform (PKM2). PKM2 expression reprograms many metabolic pathways, but little is known about its impact on cystine metabolism. Cystine metabolism is critical for supporting survival through its role in defense against ferroptosis, a non-apoptotic iron-dependent form of cell death characterized by unchecked lipid peroxidation. To improve our understanding of the role of PKM2 in cystine metabolism and ferroptosis in PDAC, we generated PKM2 knockout (KO) human PDAC cells. Fascinatingly, PKM2KO cells demonstrate a remarkable resistance to cystine starvation mediated ferroptosis. This resistance to ferroptosis is caused by decreased PK activity, rather than an isoform-specific effect. We further utilized stable isotope tracing to evaluate the impact of glucose and glutamine reprogramming in PKM2KO cells. PKM2KO cells depend on glutamine metabolism to support antioxidant defenses against lipid peroxidation, primarily by increased glutamine flux through the malate aspartate shuttle and utilization of ME1 to produce NADPH. Ferroptosis can be synergistically induced by the combination of PKM2 activation and inhibition of the cystine/glutamate antiporter in vitro. Proof-of-concept in vivo experiments demonstrate the efficacy of this mechanism as a novel treatment strategy for PDAC.
Keywords: Ferroptosis; Metabolism; Metabolomics; PKM1; PKM2; Pancreatic Cancer; Pyruvate Kinase.
Figures
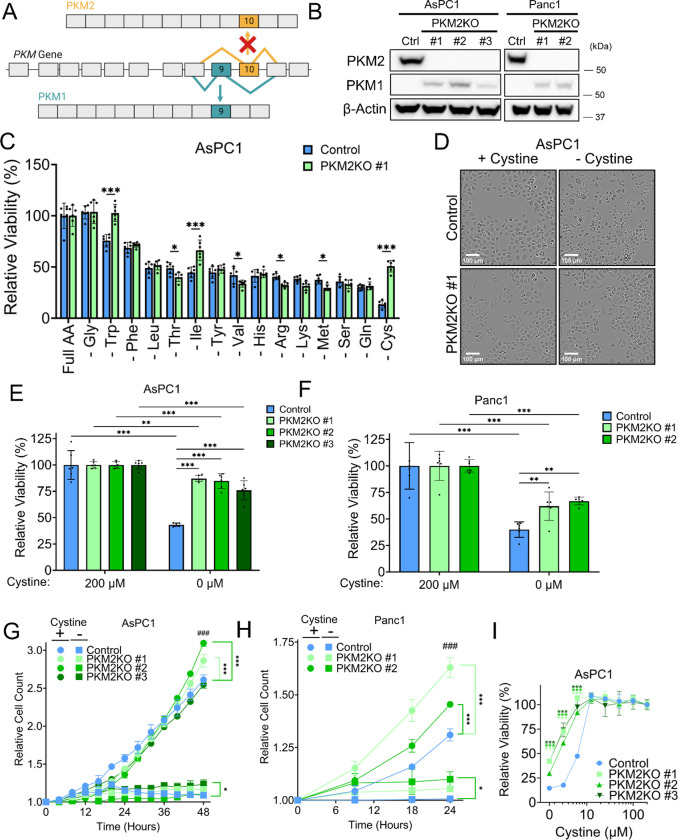
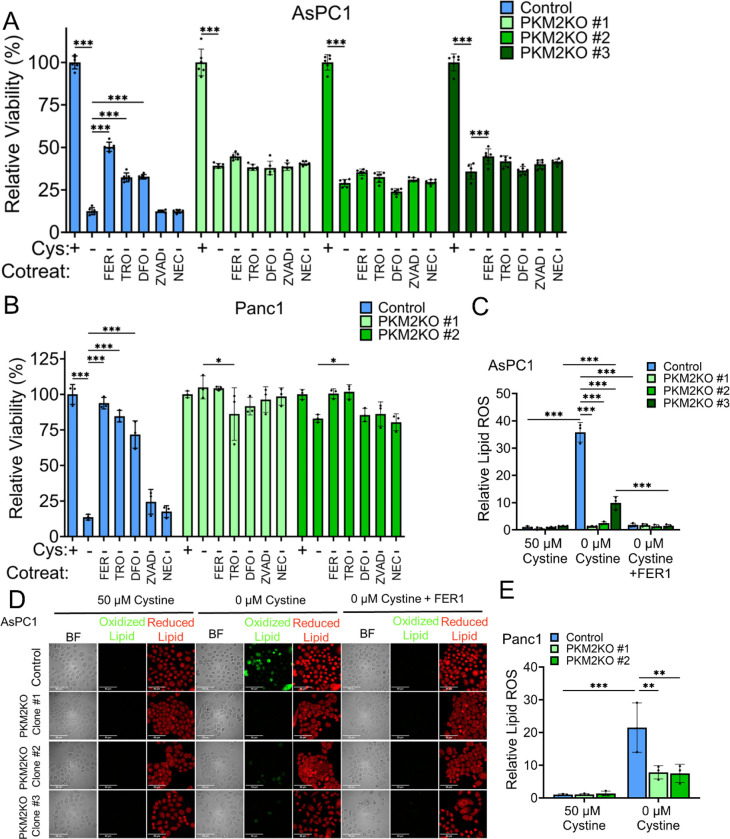
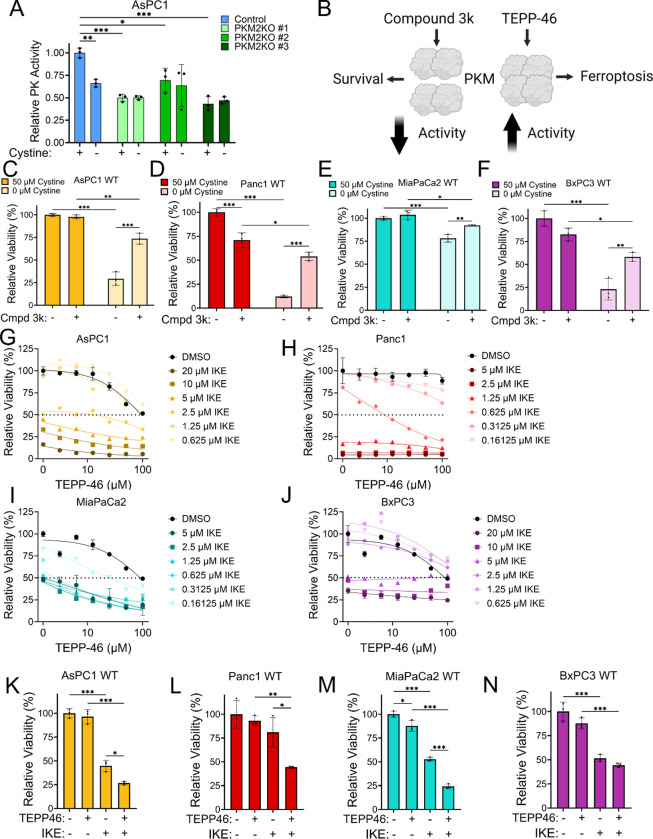
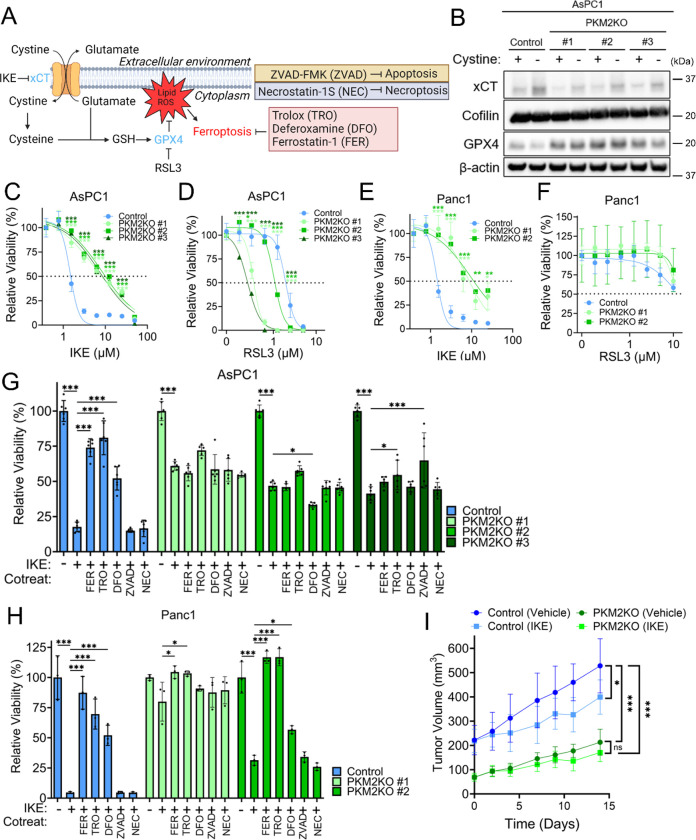
References
-
- Halbrook C. J. & Lyssiotis C. A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 31, 5–19 (2017). - PubMed
-
- Rahib L. et al. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res 74, 2913–2921 (2014). - PubMed
-
- Cancer Facts & Figures 2023. (2023).
-
- Andersson R., Pereira C.-F., Bauden M. & Ansari D. Is immunotherapy the holy grail for pancreatic cancer? Immunotherapy 11, 1435–1438 (2019). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous