

This is a preprint.
Global impact of aberrant splicing on human gene expression levels
- PMID: 37745605
- PMCID: PMC10515962
- DOI: 10.1101/2023.09.13.557588
Global impact of aberrant splicing on human gene expression levels
Update in
-
Global impact of unproductive splicing on human gene expression.Nat Genet. 2024 Sep;56(9):1851-1861. doi: 10.1038/s41588-024-01872-x. Epub 2024 Sep 2. Nat Genet. 2024. PMID: 39223315 Free PMC article.
Abstract
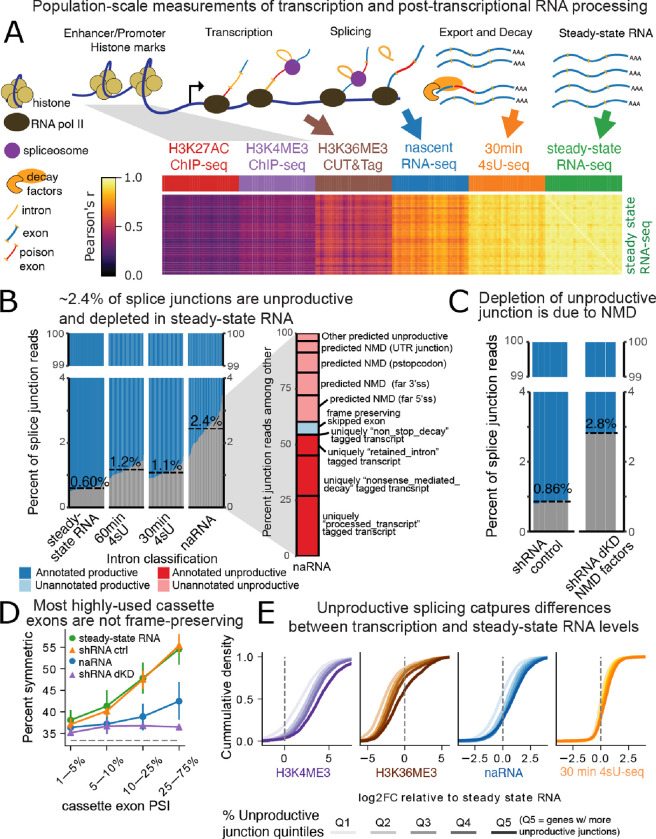
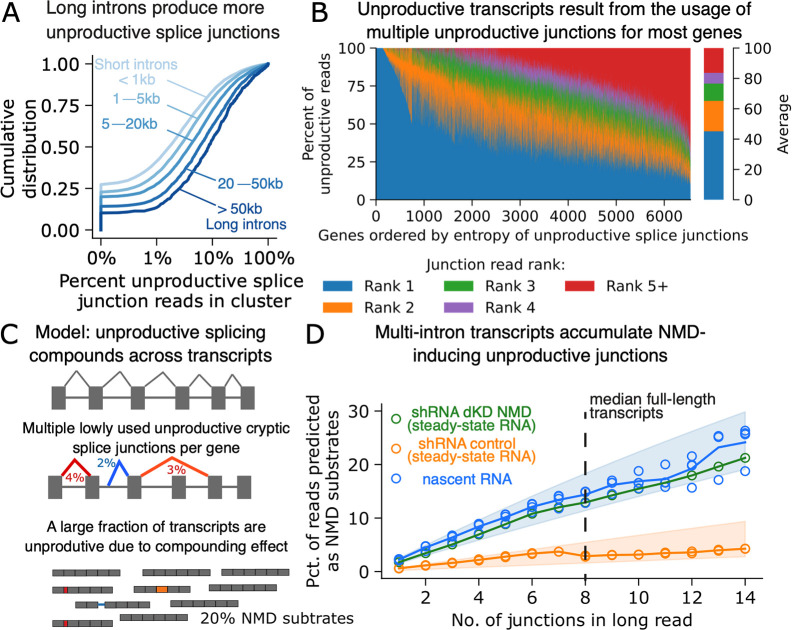
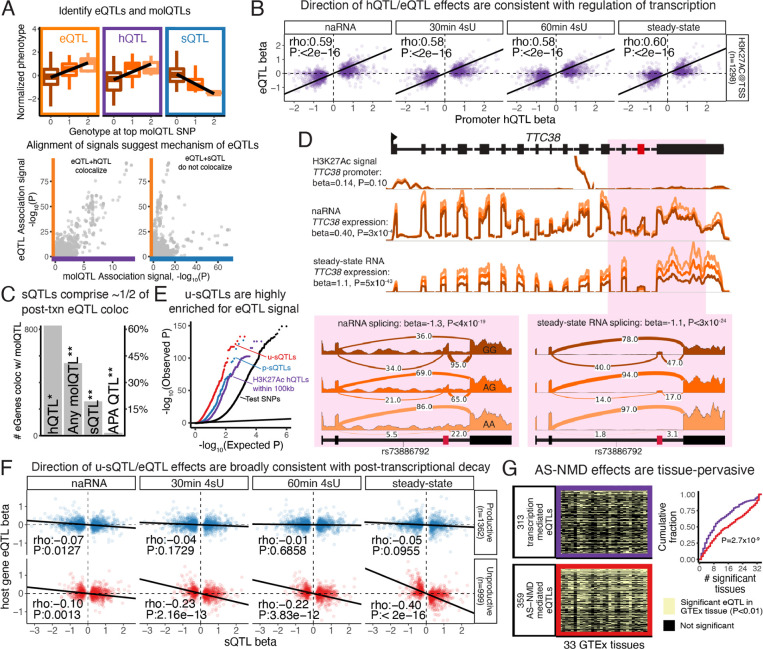
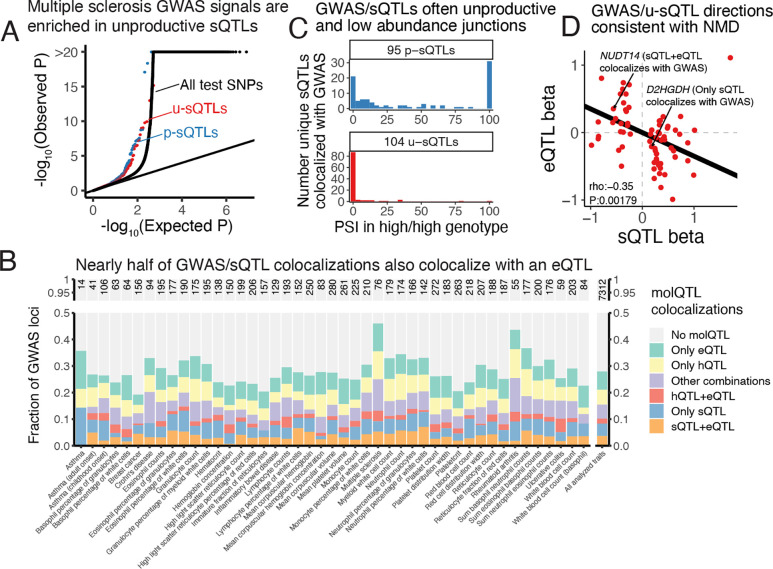
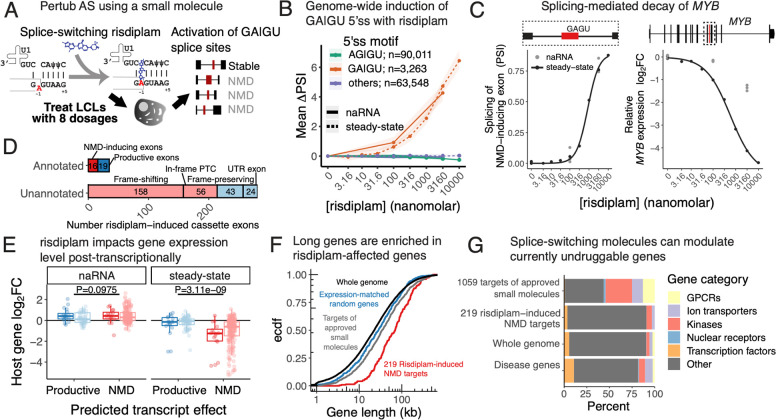
Alternative splicing (AS) is pervasive in human genes, yet the specific function of most AS events remains unknown. It is widely assumed that the primary function of AS is to diversify the proteome, however AS can also influence gene expression levels by producing transcripts rapidly degraded by nonsense-mediated decay (NMD). Currently, there are no precise estimates for how often the coupling of AS and NMD (AS-NMD) impacts gene expression levels because rapidly degraded NMD transcripts are challenging to capture. To better understand the impact of AS on gene expression levels, we analyzed population-scale genomic data in lymphoblastoid cell lines across eight molecular assays that capture gene regulation before, during, and after transcription and cytoplasmic decay. Sequencing nascent mRNA transcripts revealed frequent aberrant splicing of human introns, which results in remarkably high levels of mRNA transcripts subject to NMD. We estimate that ~15% of all protein-coding transcripts are degraded by NMD, and this estimate increases to nearly half of all transcripts for lowly-expressed genes with many introns. Leveraging genetic variation across cell lines, we find that GWAS trait-associated loci explained by AS are similarly likely to associate with NMD-induced expression level differences as with differences in protein isoform usage. Additionally, we used the splice-switching drug risdiplam to perturb AS at hundreds of genes, finding that ~3/4 of the splicing perturbations induce NMD. Thus, we conclude that AS-NMD substantially impacts the expression levels of most human genes. Our work further suggests that much of the molecular impact of AS is mediated by changes in protein expression levels rather than diversification of the proteome.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials