

Establishing induced pluripotent stem cell lines from two dominant optic atrophy patients with distinct OPA1 mutations and clinical pathologies
- PMID: 37745862
- PMCID: PMC10513078
- DOI: 10.3389/fgene.2023.1251216
Establishing induced pluripotent stem cell lines from two dominant optic atrophy patients with distinct OPA1 mutations and clinical pathologies
Abstract
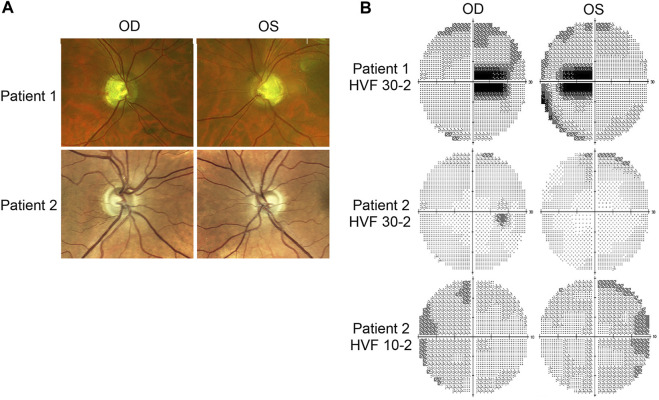
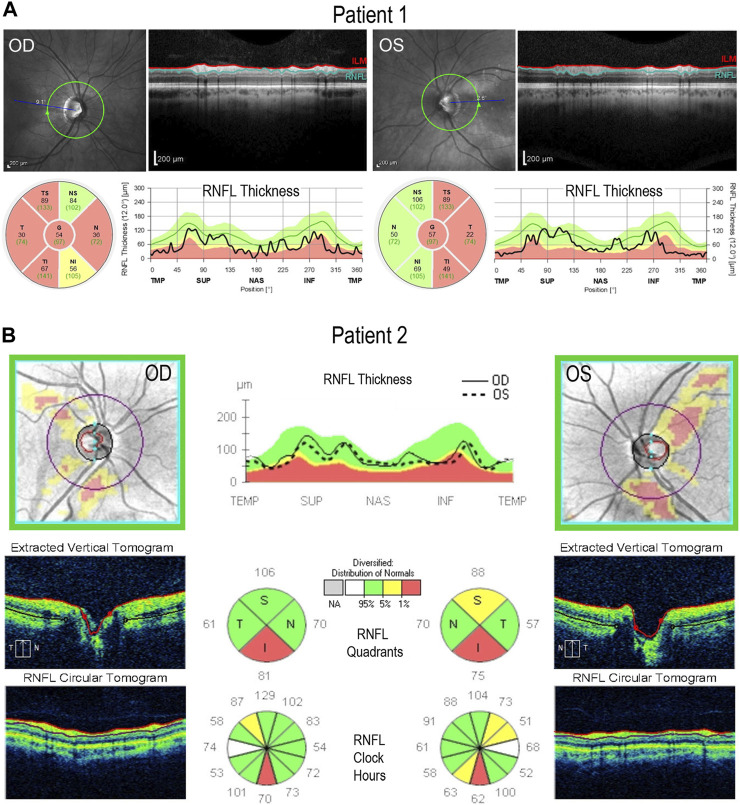
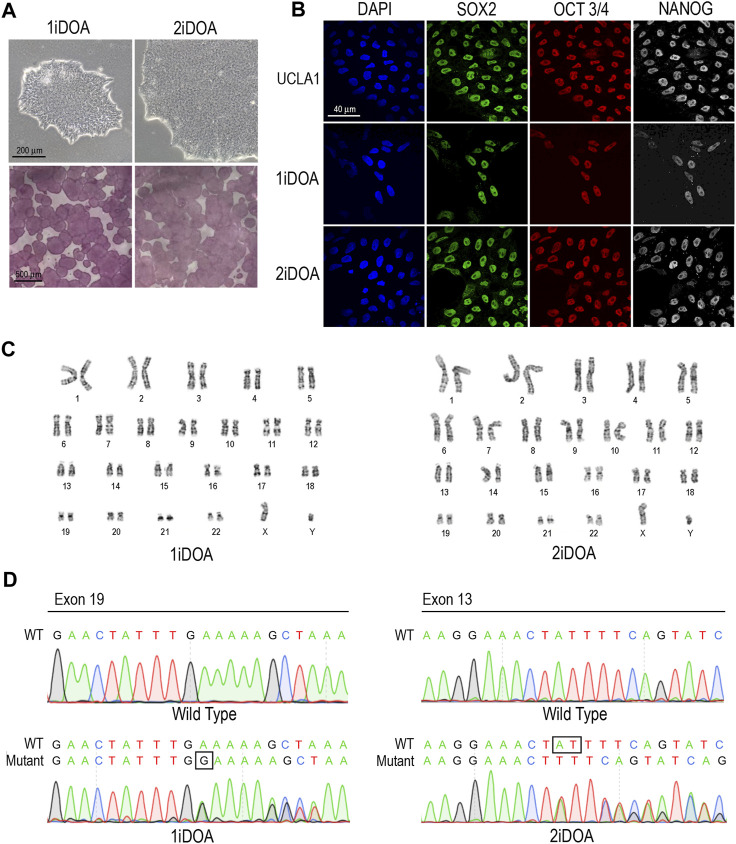
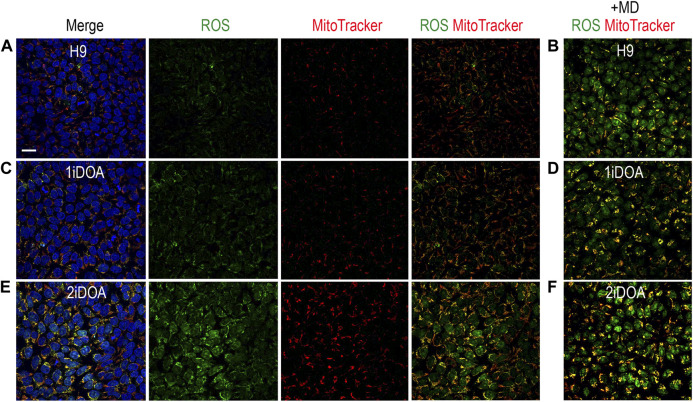
Dominant optic atrophy (DOA) is an inherited disease that leads to the loss of retinal ganglion cells (RGCs), the projection neurons that relay visual information from the retina to the brain through the optic nerve. The majority of DOA cases can be attributed to mutations in optic atrophy 1 (OPA1), a nuclear gene encoding a mitochondrial-targeted protein that plays important roles in maintaining mitochondrial structure, dynamics, and bioenergetics. Although OPA1 is ubiquitously expressed in all human tissues, RGCs appear to be the primary cell type affected by OPA1 mutations. DOA has not been extensively studied in human RGCs due to the general unavailability of retinal tissues. However, recent advances in stem cell biology have made it possible to produce human RGCs from pluripotent stem cells (PSCs). To aid in establishing DOA disease models based on human PSC-derived RGCs, we have generated iPSC lines from two DOA patients who carry distinct OPA1 mutations and present very different disease symptoms. Studies using these OPA1 mutant RGCs can be correlated with clinical features in the patients to provide insights into DOA disease mechanisms.
Keywords: DOA; OPA1; RGC; dominant optic atrophy; induced pluripotent stem cells (iPSC); retinal ganglion cell.
Copyright © 2023 Pohl, Zhang, Pham, Chan, Sadun and Yang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Agier V., Oliviero P., Laine J., L'Hermitte-Stead C., Girard S., Fillaut S., et al. (2012). Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim. Biophys. Acta 1822 (10), 1570–1580. 10.1016/j.bbadis.2012.07.002 - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources