

Tertiary structure assessment at CASP15
- PMID: 37746927
- PMCID: PMC10792517
- DOI: 10.1002/prot.26593
Tertiary structure assessment at CASP15
Abstract
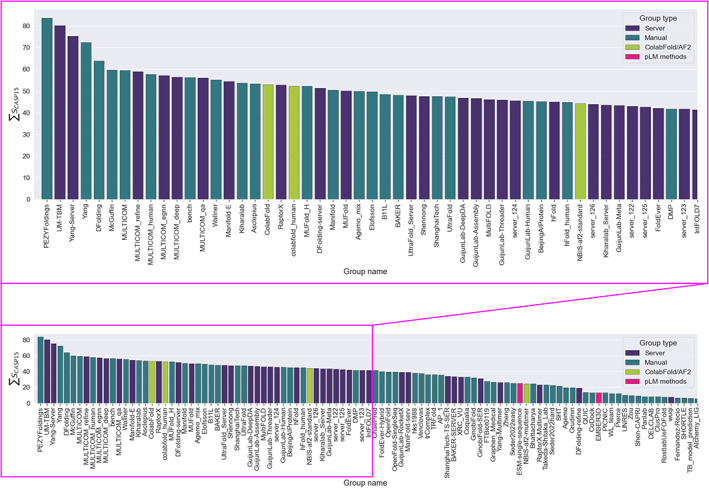
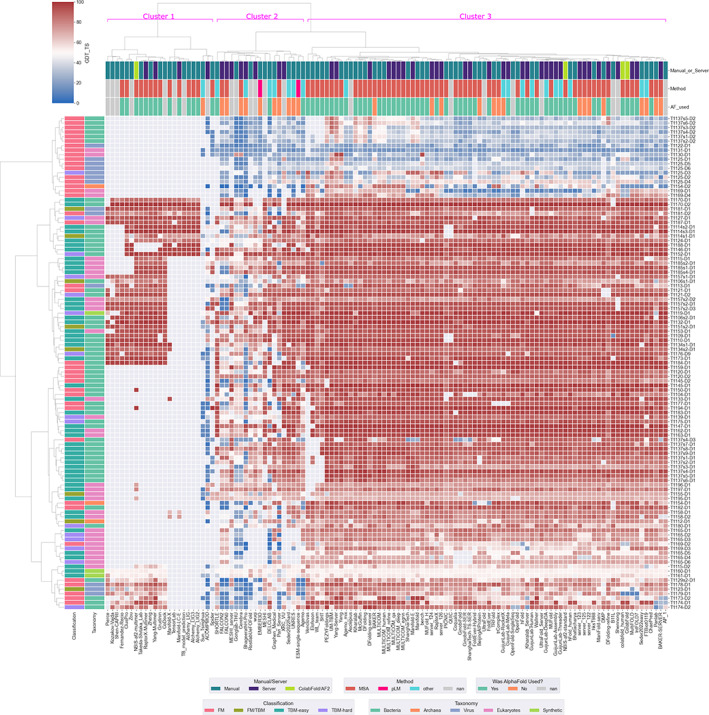
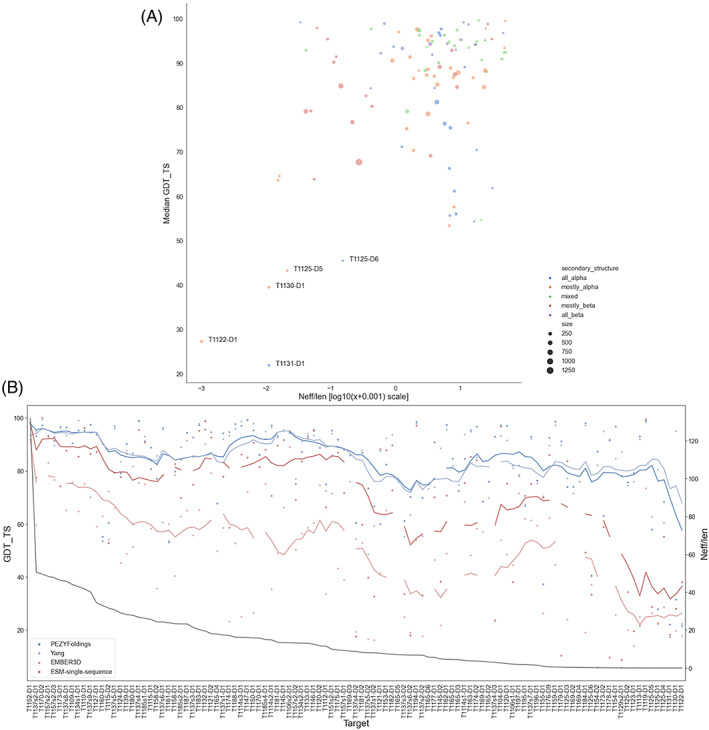
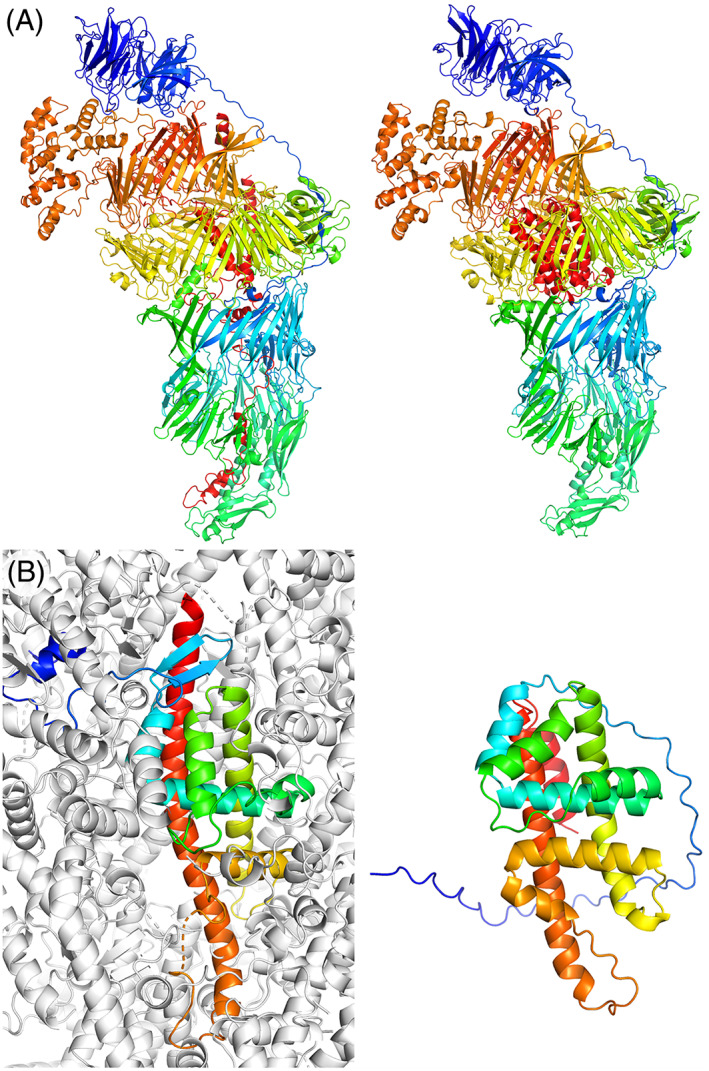
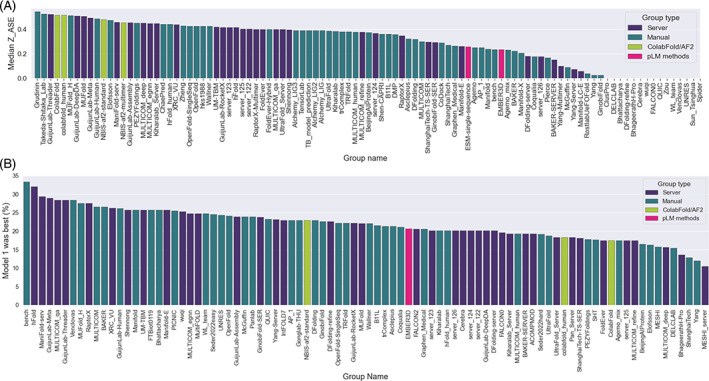
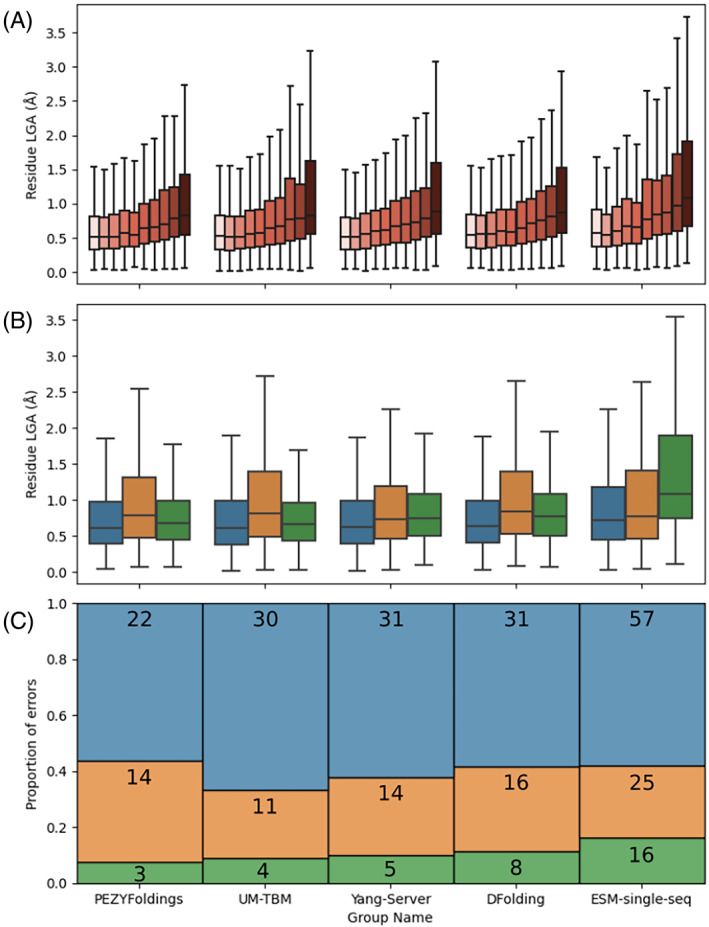
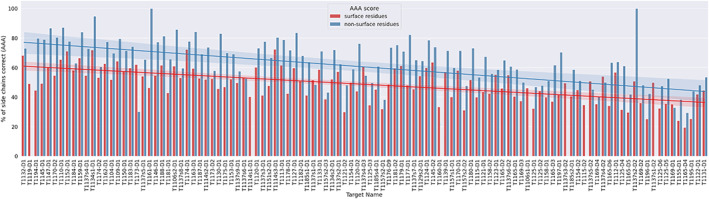
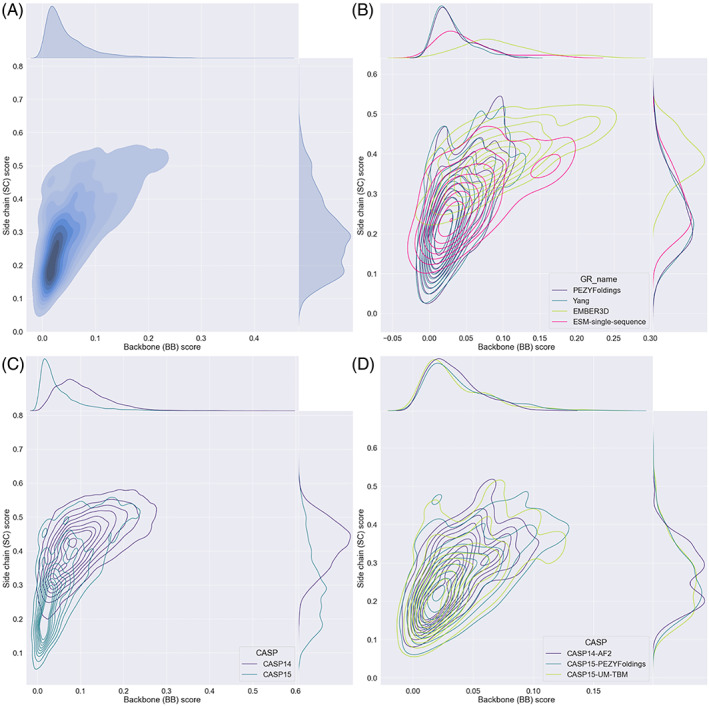
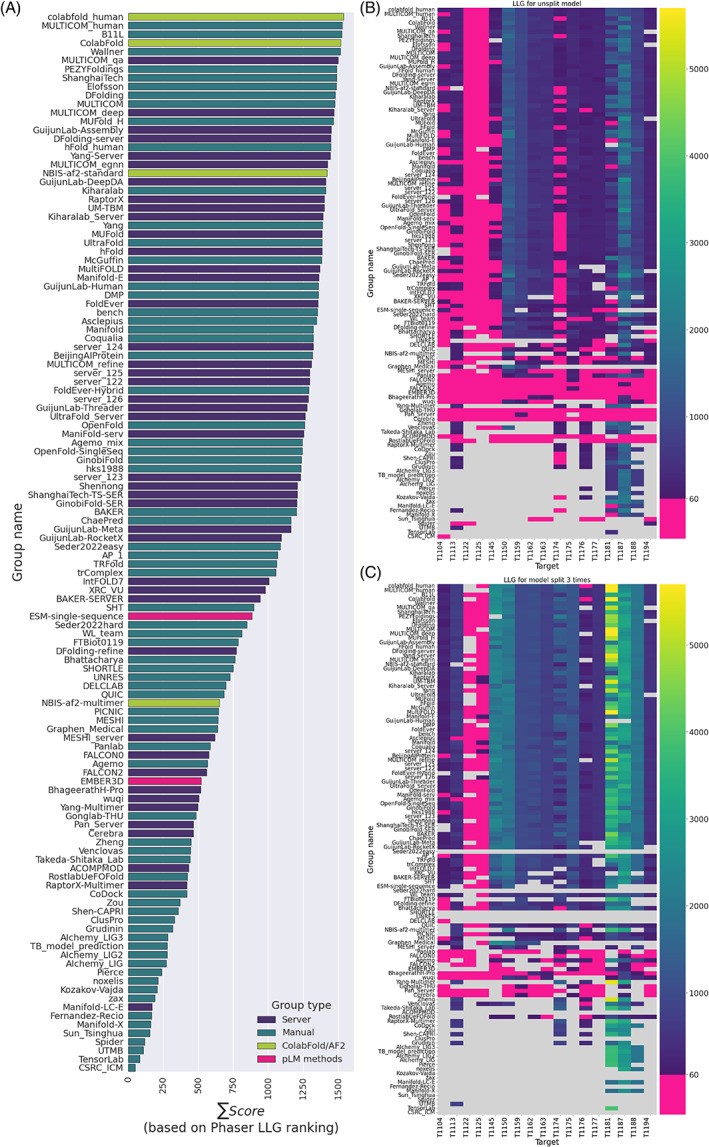
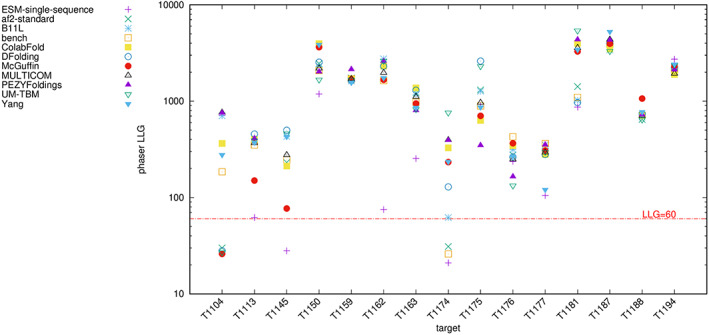
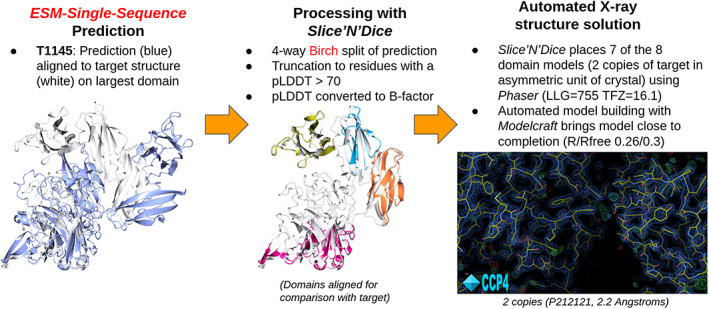
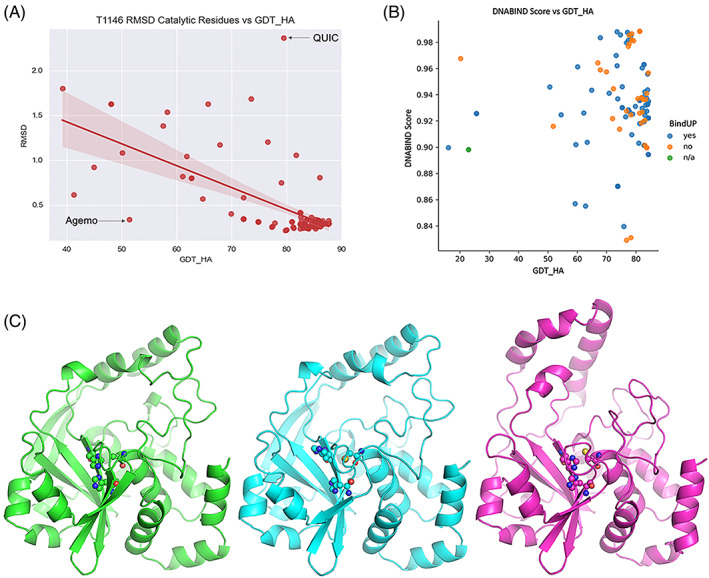
The results of tertiary structure assessment at CASP15 are reported. For the first time, recognizing the outstanding performance of AlphaFold 2 (AF2) at CASP14, all single-chain predictions were assessed together, irrespective of whether a template was available. At CASP15, there was no single stand-out group, with most of the best-scoring groups-led by PEZYFoldings, UM-TBM, and Yang Server-employing AF2 in one way or another. Many top groups paid special attention to generating deep Multiple Sequence Alignments (MSAs) and testing variant MSAs, thereby allowing them to successfully address some of the hardest targets. Such difficult targets, as well as lacking templates, were typically proteins with few homologues. Local divergence between prediction and target correlated with localization at crystal lattice or chain interfaces, and with regions exhibiting high B-factor factors in crystal structure targets, and should not necessarily be considered as representing error in the prediction. However, analysis of exposed and buried side chain accuracy showed room for improvement even in the latter. Nevertheless, a majority of groups produced high-quality predictions for most targets, which are valuable for experimental structure determination, functional analysis, and many other tasks across biology. These include those applying methods similar to those used to generate major resources such as the AlphaFold Protein Structure Database and the ESM Metagenomic atlas: the confidence estimates of the former were also notably accurate.
Keywords: CASP15; machine learning; molecular replacement; protein modelling; protein structure prediction; structural bioinformatics.
© 2023 The Authors. Proteins: Structure, Function, and Bioinformatics published by Wiley Periodicals LLC.
Figures
References
-
- Moult J, Pedersen JT, Judson R, Fidelis K. A large‐scale experiment to assess protein structure prediction methods. Proteins. 1995;23(3):ii‐v. - PubMed
-
- Burley SK, Bhikadiya C, Bi C, et al. RCSB protein data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021;49(D1):D437‐D451. - PMC - PubMed
-
- Defay T, Cohen FE. Evaluation of current techniques for ab initio protein structure prediction. Proteins. 1995;23(3):431‐445. - PubMed
