

Driving forces behind phase separation of the carboxy-terminal domain of RNA polymerase II
- PMID: 37749095
- PMCID: PMC10519987
- DOI: 10.1038/s41467-023-41633-8
Driving forces behind phase separation of the carboxy-terminal domain of RNA polymerase II
Abstract
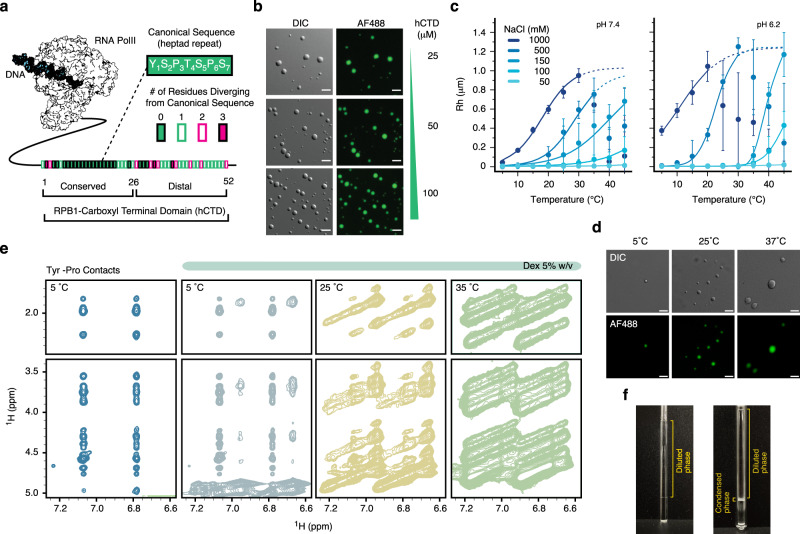
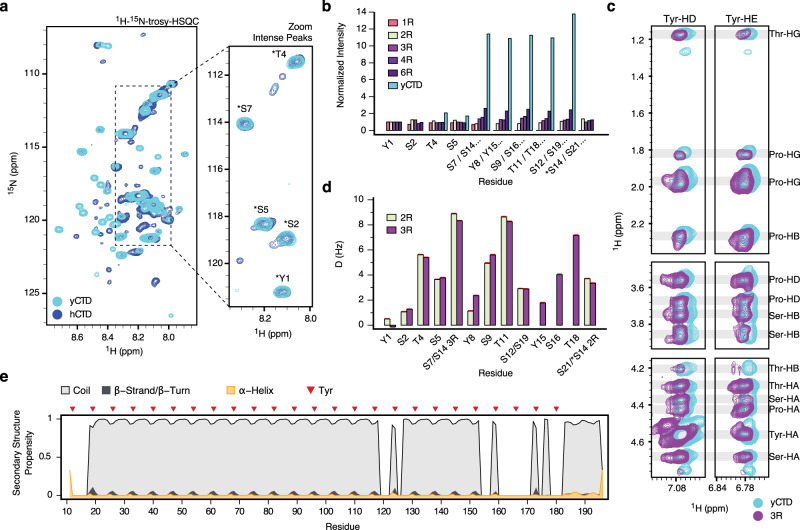
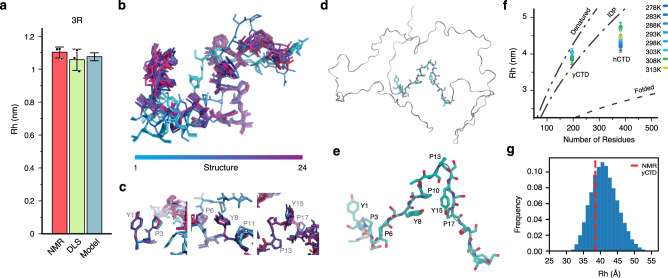
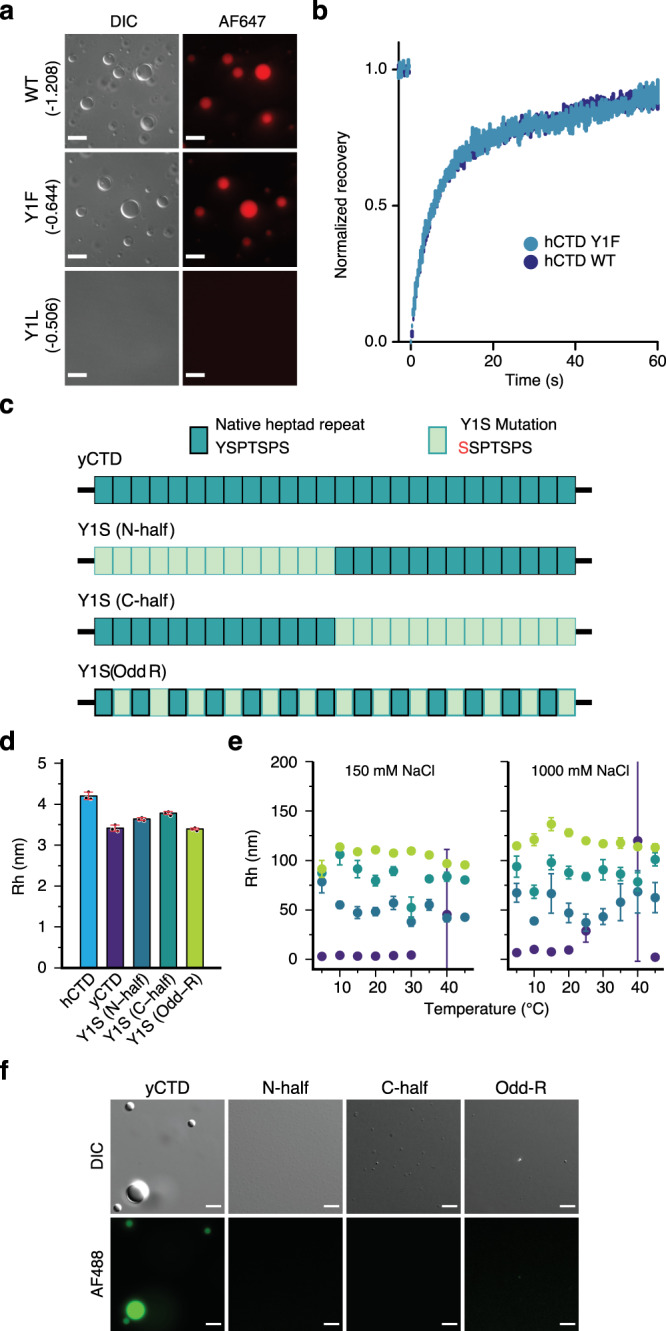
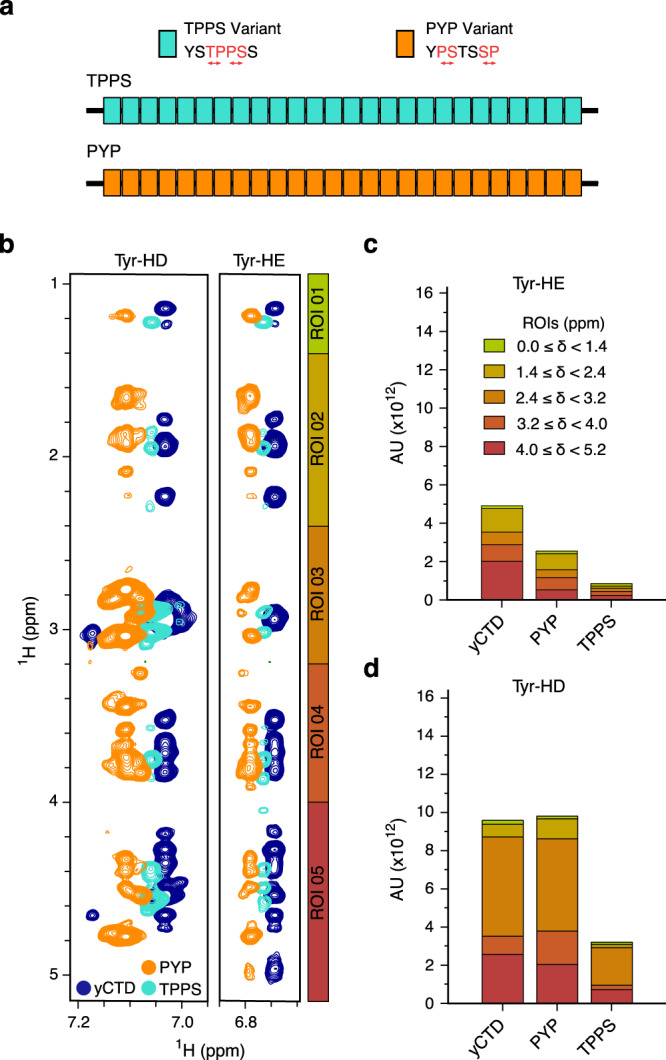
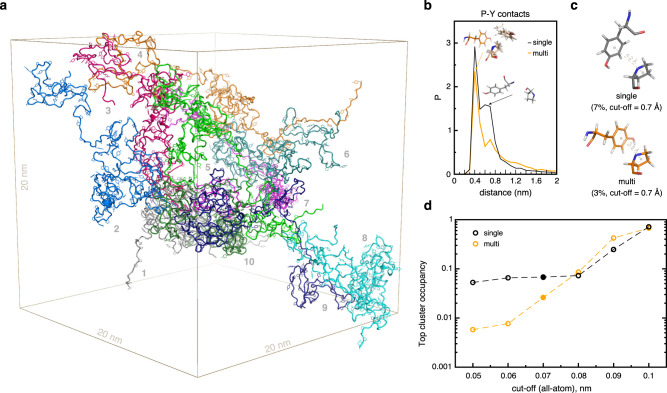
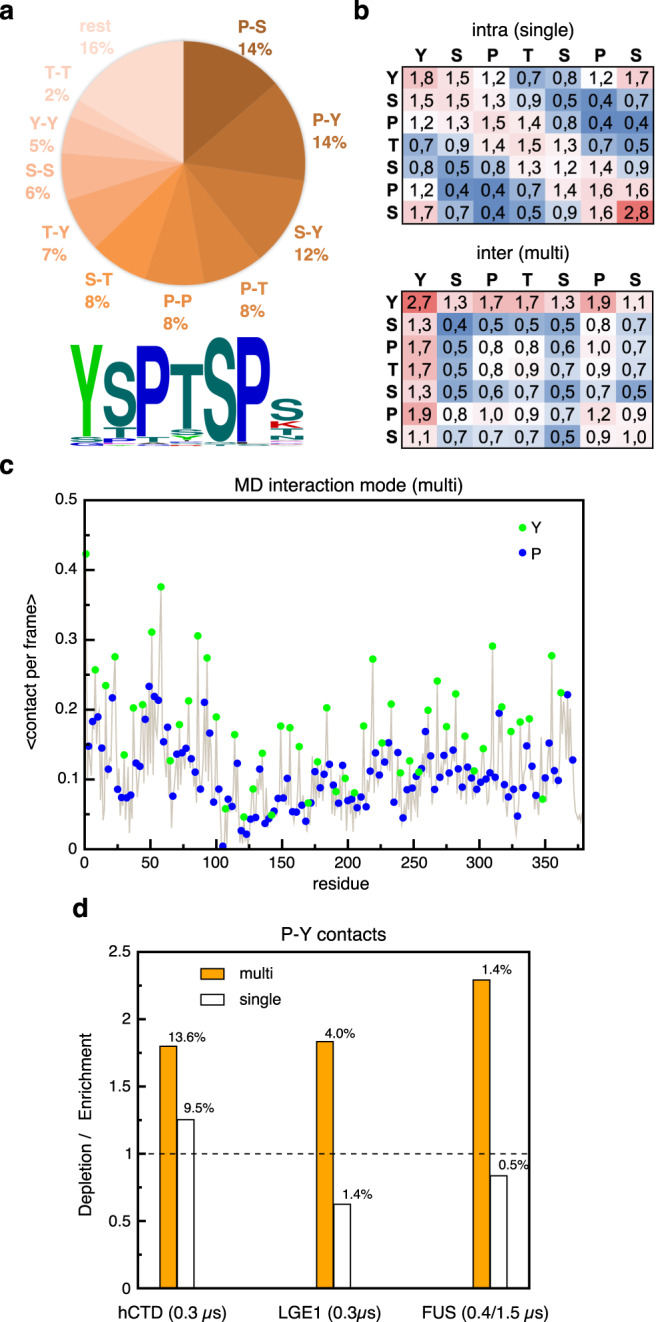
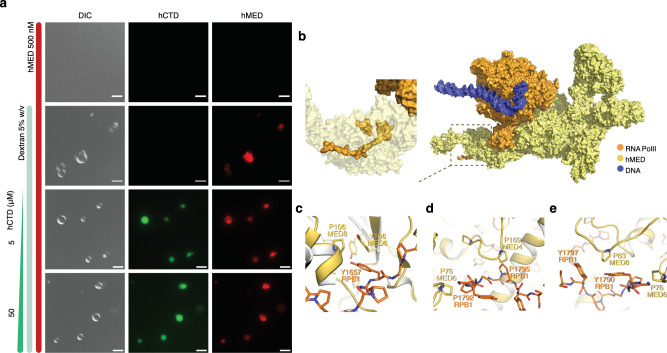
Eukaryotic gene regulation and pre-mRNA transcription depend on the carboxy-terminal domain (CTD) of RNA polymerase (Pol) II. Due to its highly repetitive, intrinsically disordered sequence, the CTD enables clustering and phase separation of Pol II. The molecular interactions that drive CTD phase separation and Pol II clustering are unclear. Here, we show that multivalent interactions involving tyrosine impart temperature- and concentration-dependent self-coacervation of the CTD. NMR spectroscopy, molecular ensemble calculations and all-atom molecular dynamics simulations demonstrate the presence of diverse tyrosine-engaging interactions, including tyrosine-proline contacts, in condensed states of human CTD and other low-complexity proteins. We further show that the network of multivalent interactions involving tyrosine is responsible for the co-recruitment of the human Mediator complex and CTD during phase separation. Our work advances the understanding of the driving forces of CTD phase separation and thus provides the basis to better understand CTD-mediated Pol II clustering in eukaryotic gene transcription.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
RNA polymerase II clustering through carboxy-terminal domain phase separation.Nat Struct Mol Biol. 2018 Sep;25(9):833-840. doi: 10.1038/s41594-018-0112-y. Epub 2018 Aug 20. Nat Struct Mol Biol. 2018. PMID: 30127355
-
The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain.Nat Rev Mol Cell Biol. 2017 Apr;18(4):263-273. doi: 10.1038/nrm.2017.10. Epub 2017 Mar 1. Nat Rev Mol Cell Biol. 2017. PMID: 28248323 Review.
-
Structure and phase separation of the C-terminal domain of RNA polymerase II.Biol Chem. 2023 Jun 20;404(8-9):839-844. doi: 10.1515/hsz-2023-0136. Print 2023 Jul 26. Biol Chem. 2023. PMID: 37331973
-
Removing quote marks from the RNA polymerase II CTD 'code'.Biosystems. 2021 Sep;207:104468. doi: 10.1016/j.biosystems.2021.104468. Epub 2021 Jun 30. Biosystems. 2021. PMID: 34216714
-
The carboxy-terminal domain of RNA polymerase II large subunit: simple repeats are not simple.Yi Chuan. 2024 Dec;46(12):1028-1041. doi: 10.16288/j.yczz.24-200. Yi Chuan. 2024. PMID: 39658958 Review.
Cited by
-
Gene dosage and protein valency impact phase separation and fungal cell fate.PLoS Genet. 2025 Aug 8;21(8):e1011810. doi: 10.1371/journal.pgen.1011810. eCollection 2025 Aug. PLoS Genet. 2025. PMID: 40779608 Free PMC article.
-
Variation of C-terminal domain governs RNA polymerase II genomic locations and alternative splicing in eukaryotic transcription.bioRxiv [Preprint]. 2024 Jan 2:2024.01.01.573828. doi: 10.1101/2024.01.01.573828. bioRxiv. 2024. Update in: Nat Commun. 2024 Sep 12;15(1):7985. doi: 10.1038/s41467-024-52391-6. PMID: 38260389 Free PMC article. Updated. Preprint.
-
Phosphorylation modulates secondary structure of intrinsically disorder regions in RNA polymerase II.J Biol Chem. 2025 Jun;301(6):108533. doi: 10.1016/j.jbc.2025.108533. Epub 2025 Apr 22. J Biol Chem. 2025. PMID: 40273986 Free PMC article.
-
Selective phase separation of transcription factors is driven by orthogonal molecular grammar.Nat Commun. 2025 Mar 31;16(1):3087. doi: 10.1038/s41467-025-58445-7. Nat Commun. 2025. PMID: 40164612 Free PMC article.
-
Sequence and structural determinants of RNAPII CTD phase-separation and phosphorylation by CDK7.Nat Commun. 2024 Oct 24;15(1):9163. doi: 10.1038/s41467-024-53305-2. Nat Commun. 2024. PMID: 39448580 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
