

A new method for multiancestry polygenic prediction improves performance across diverse populations
- PMID: 37749244
- PMCID: PMC10923245
- DOI: 10.1038/s41588-023-01501-z
A new method for multiancestry polygenic prediction improves performance across diverse populations
Abstract
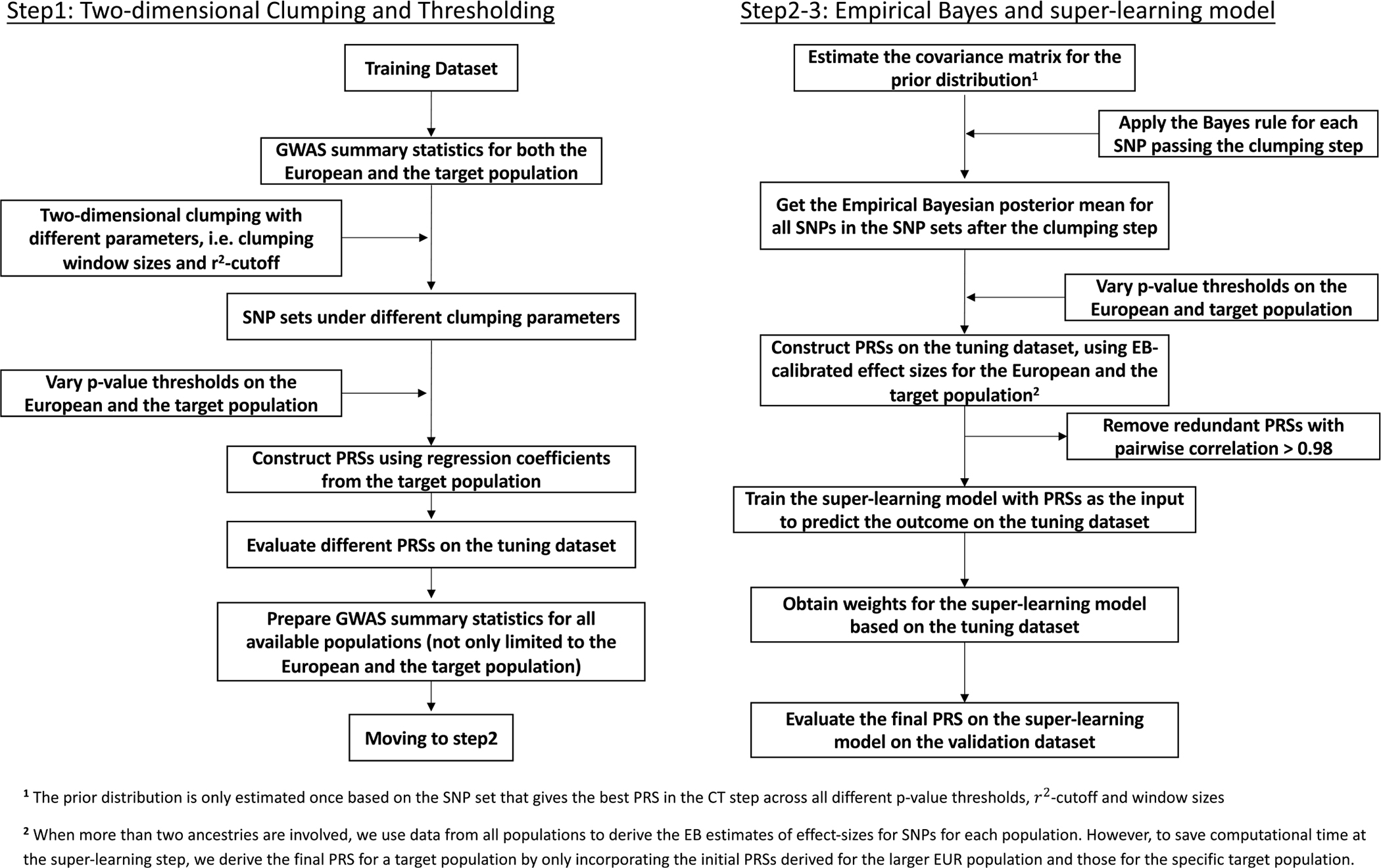
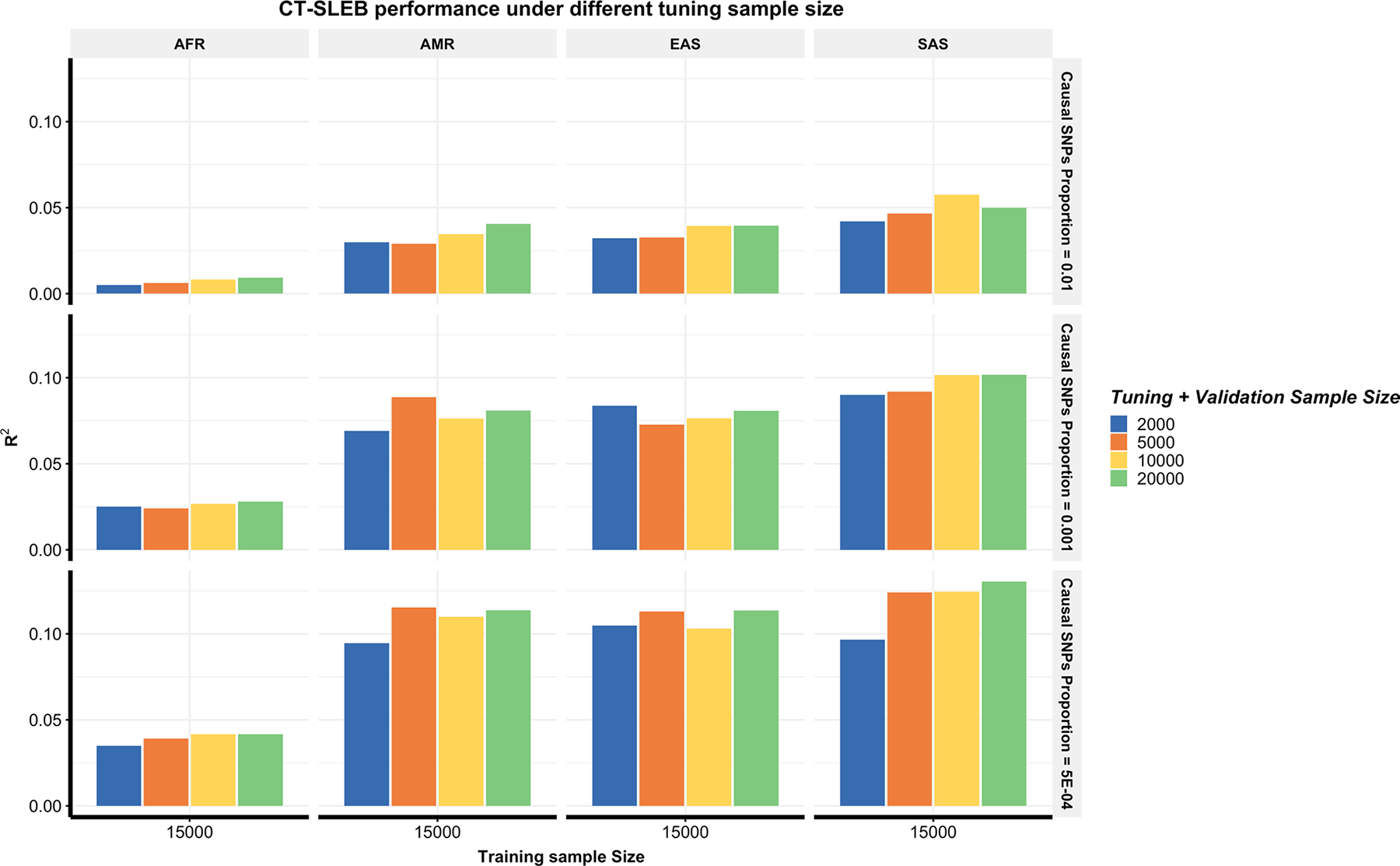
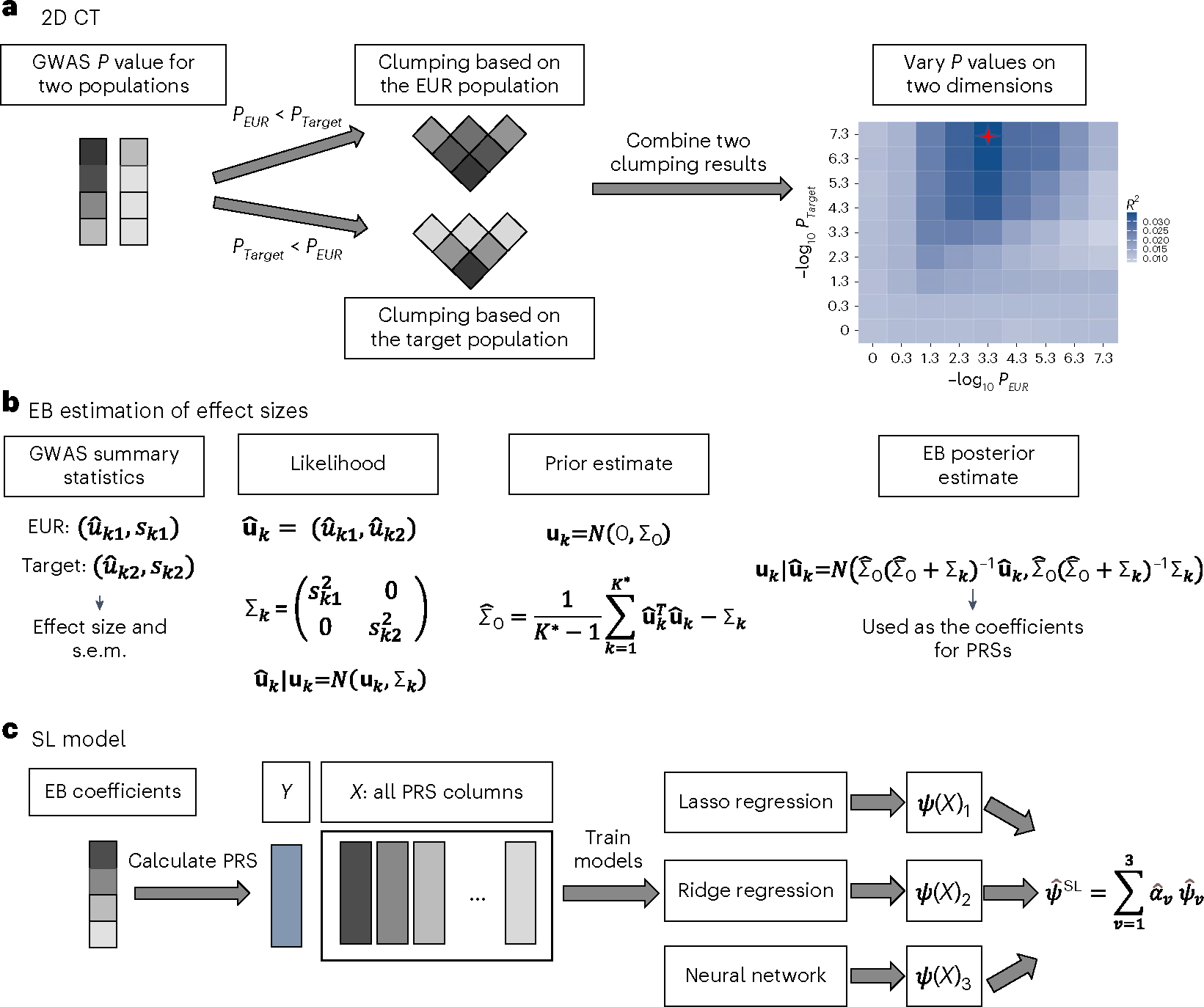
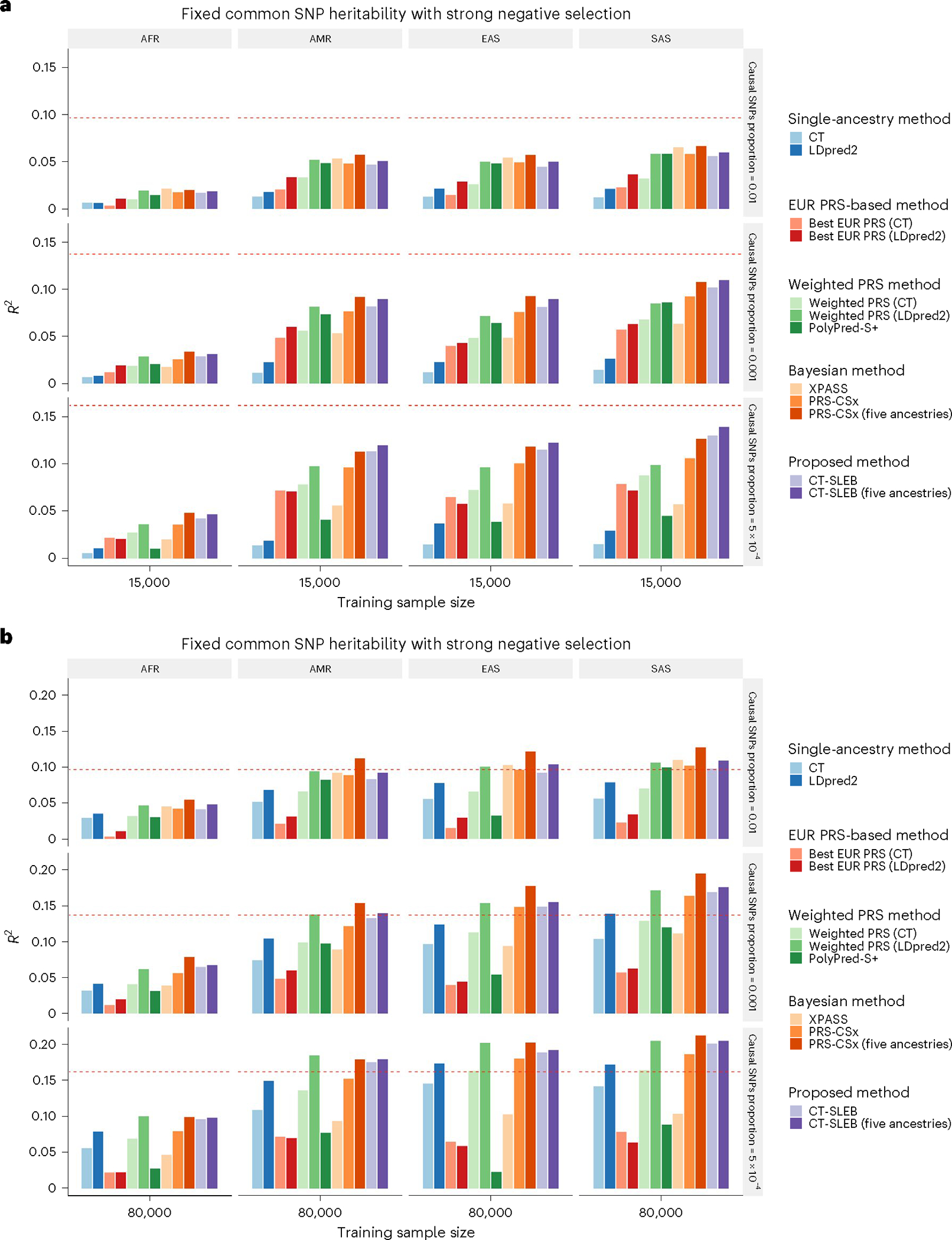
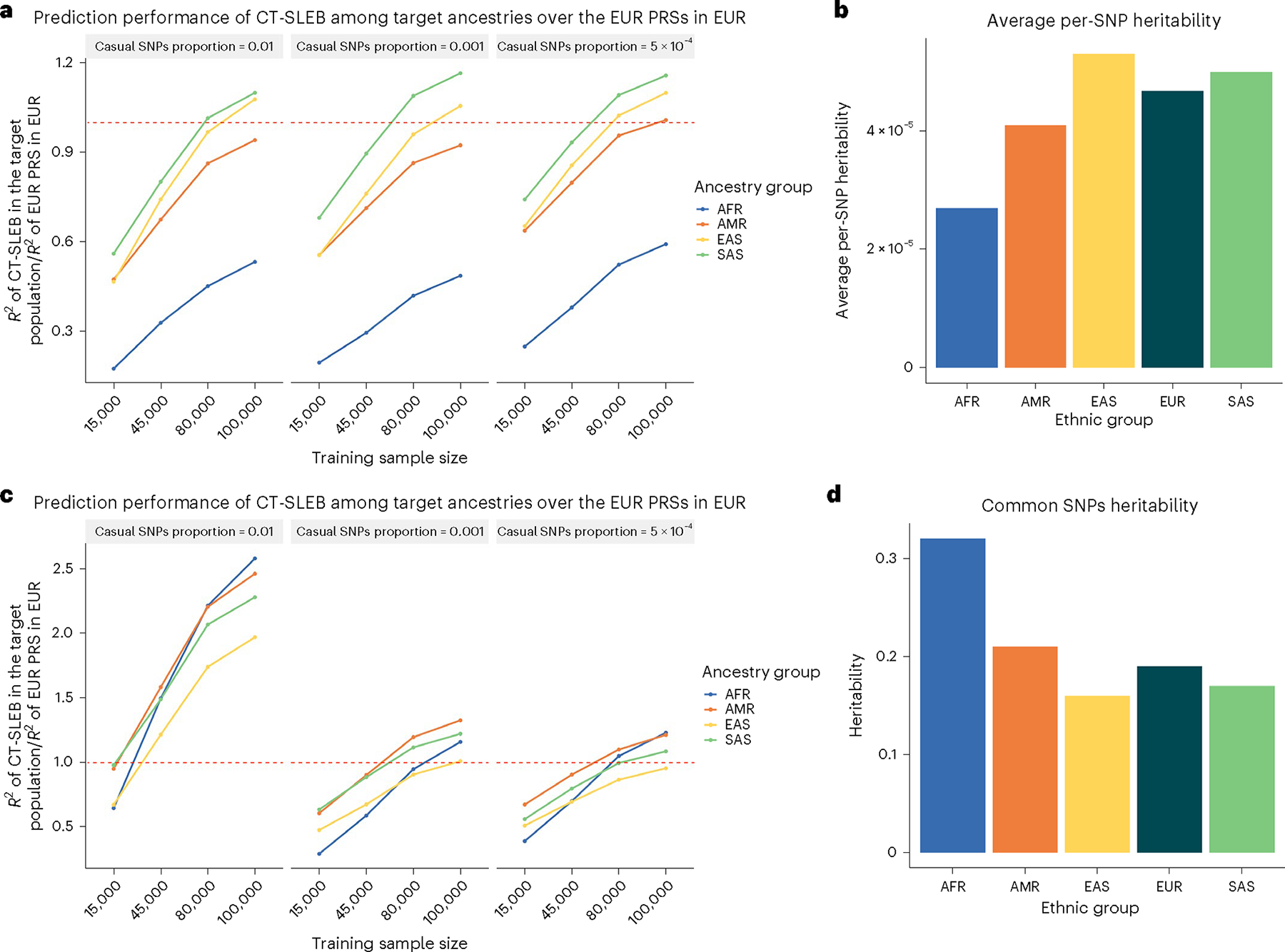
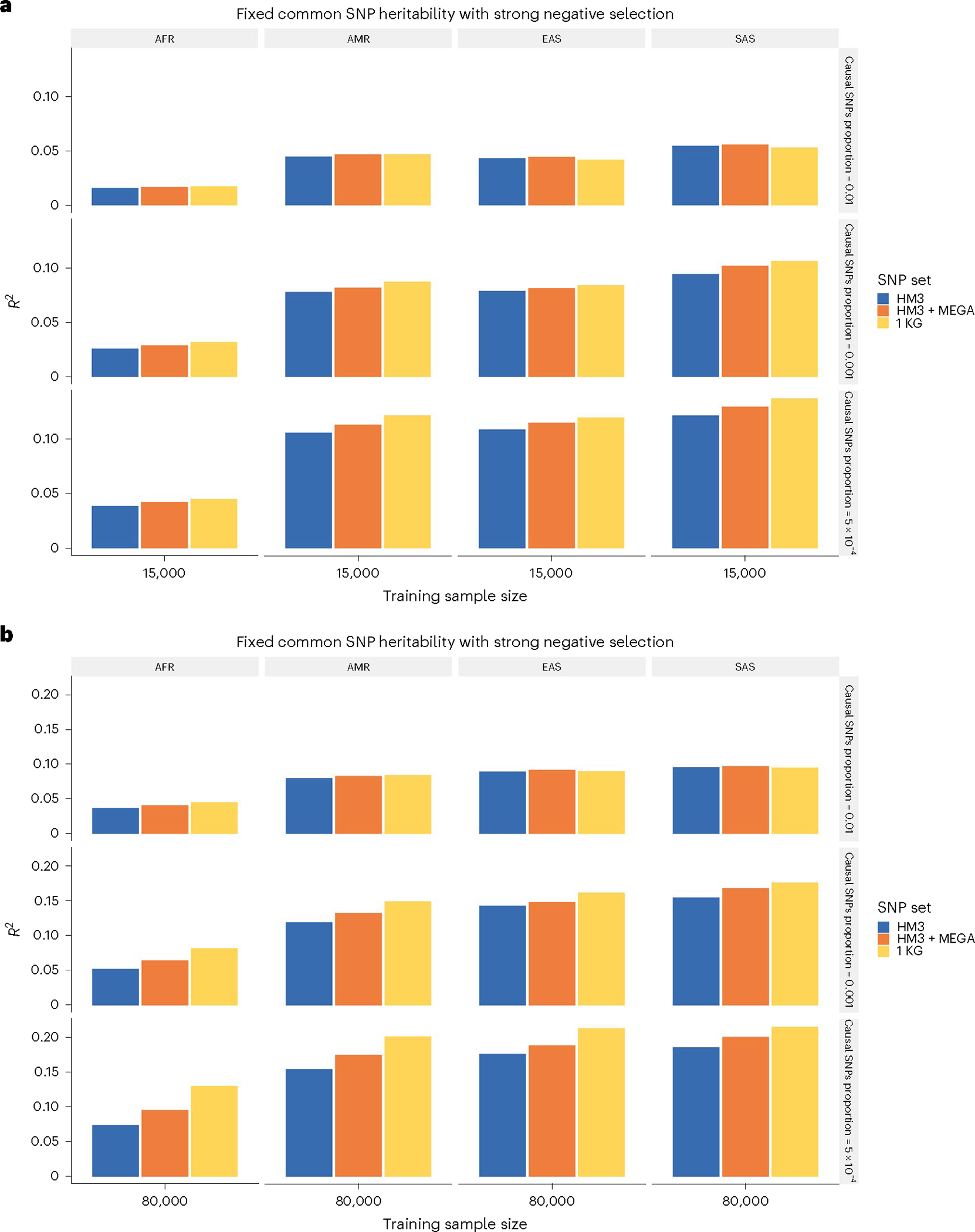
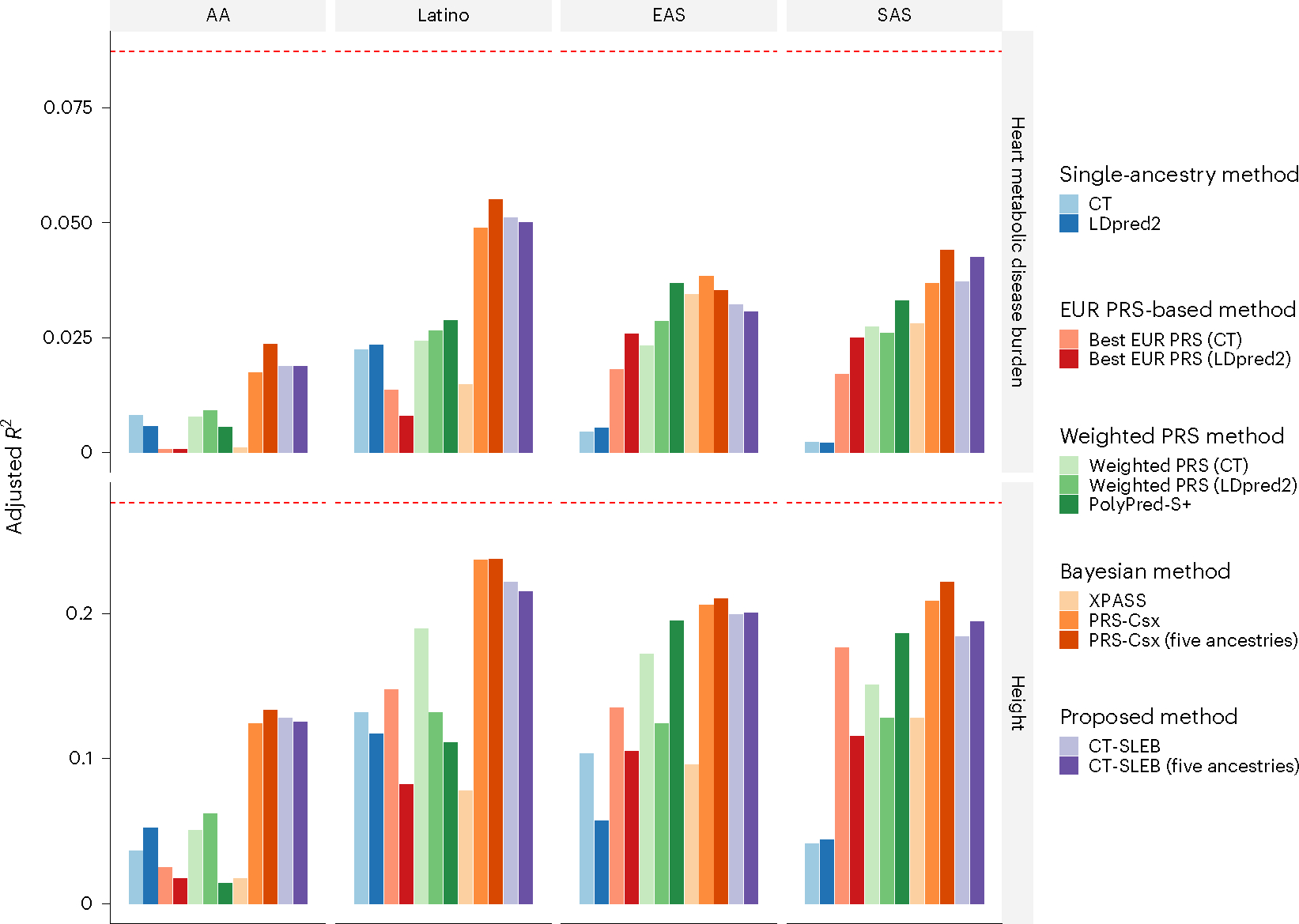
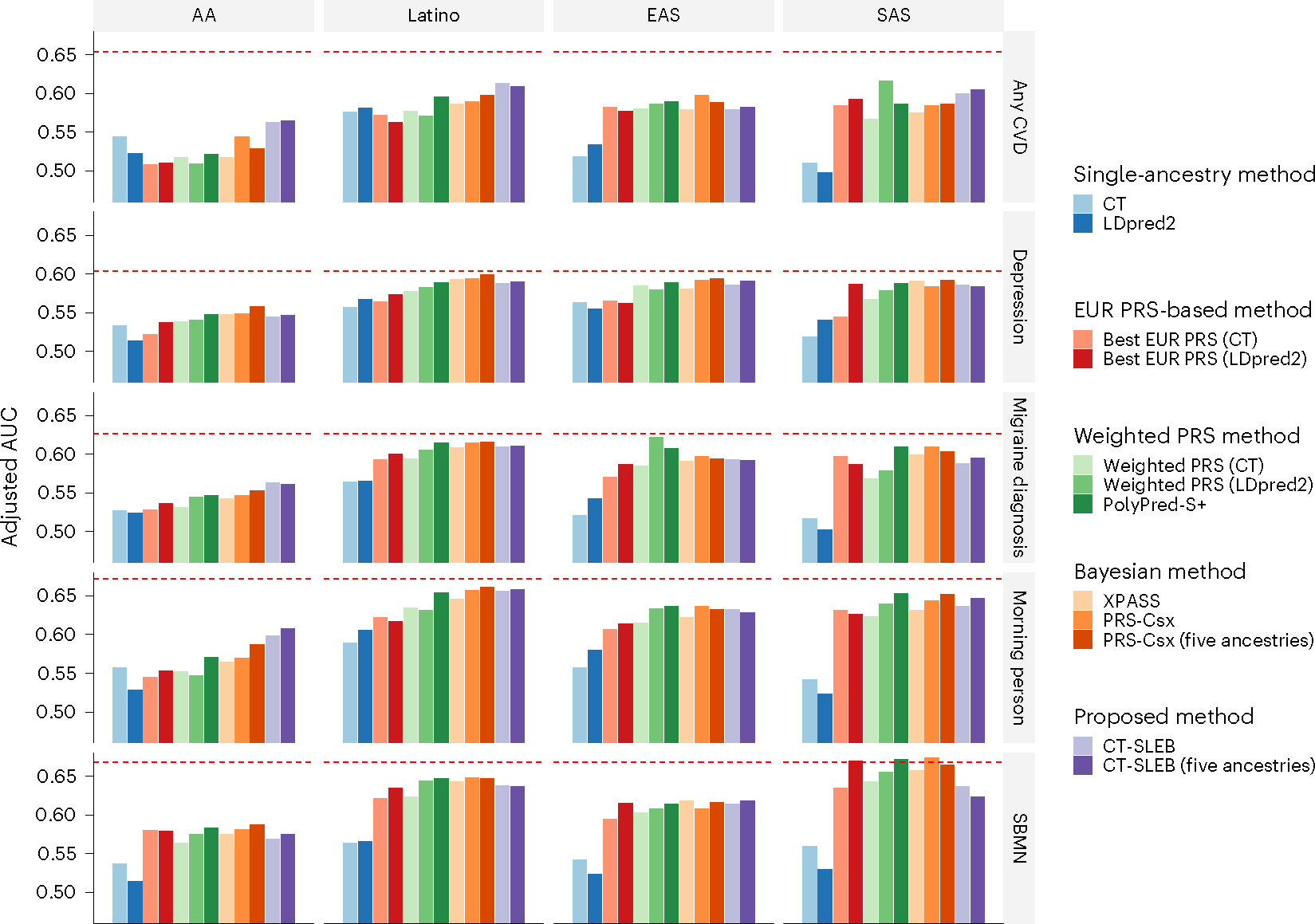
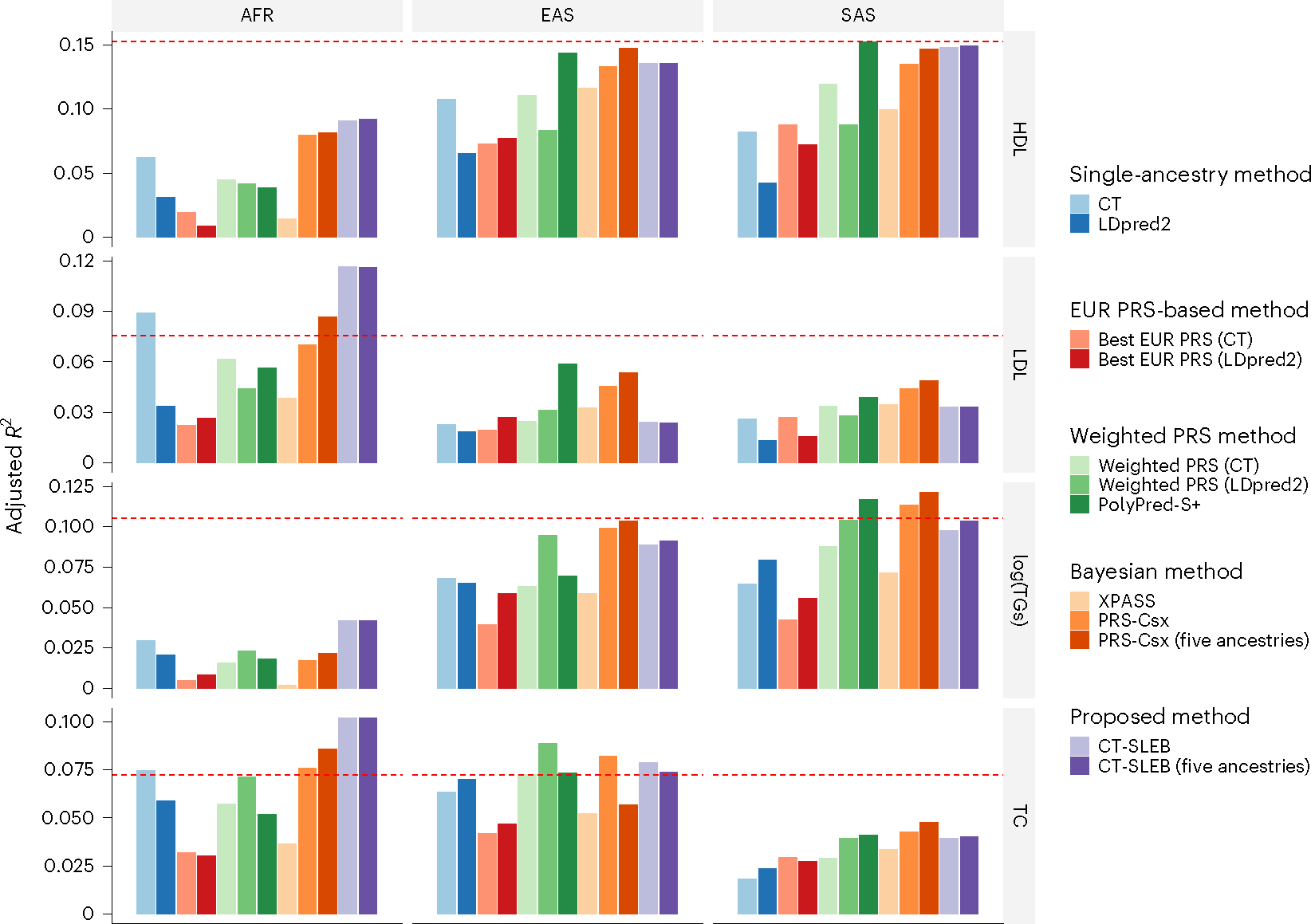
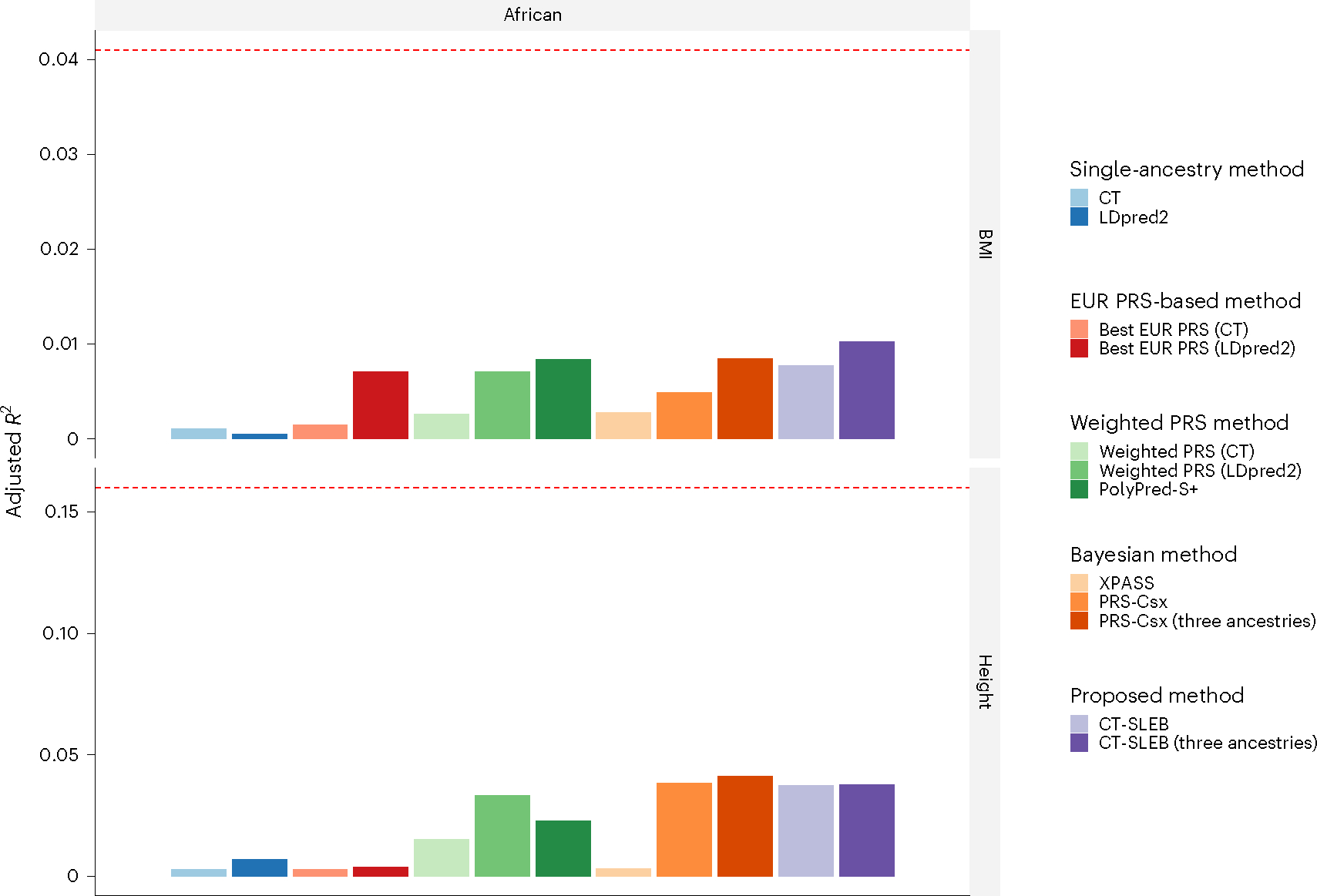
Polygenic risk scores (PRSs) increasingly predict complex traits; however, suboptimal performance in non-European populations raise concerns about clinical applications and health inequities. We developed CT-SLEB, a powerful and scalable method to calculate PRSs, using ancestry-specific genome-wide association study summary statistics from multiancestry training samples, integrating clumping and thresholding, empirical Bayes and superlearning. We evaluated CT-SLEB and nine alternative methods with large-scale simulated genome-wide association studies (~19 million common variants) and datasets from 23andMe, Inc., the Global Lipids Genetics Consortium, All of Us and UK Biobank, involving 5.1 million individuals of diverse ancestry, with 1.18 million individuals from four non-European populations across 13 complex traits. Results demonstrated that CT-SLEB significantly improves PRS performance in non-European populations compared with simple alternatives, with comparable or superior performance to a recent, computationally intensive method. Moreover, our simulation studies offered insights into sample size requirements and SNP density effects on multiancestry risk prediction.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
J.Z., J.O., Y.J., S.A., A.A., E.B., R.K.B., J.B., K.B., E.B., D.C., G.C.P., D.D., S.D., S.L.E., N.E., T.F., A.F., K.F.B., P.F., W.F., J.M.G., K.H., A.H., B.H., D.A.H., E.M.J., K.K., A.K., K.H.L., B.A.L., M.L., J.C.M., M.H.M., S.J.M., M.E.M., P.N., D.T.N., E.S.N., A.A.P., G.D.P., A.R., M.S., A.J.S., J.F.S., J.S., S.S., Q.J.S., S.A.T., C.T.T., V.T., J.Y.T., X.W., W.W., C.H.W., P.W., C.D.W. and B.L.K. are employed by and hold stock or stock options in 23andMe, Inc. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- R35 CA197449/CA/NCI NIH HHS/United States
- U01 HG012064/HG/NHGRI NIH HHS/United States
- U2C OD023196/OD/NIH HHS/United States
- OT2 OD026551/OD/NIH HHS/United States
- U24 OD023121/OD/NIH HHS/United States
- OT2 OD026549/OD/NIH HHS/United States
- OT2 OD025337/OD/NIH HHS/United States
- OT2 OD025277/OD/NIH HHS/United States
- OT2 OD026555/OD/NIH HHS/United States
- OT2 OD026550/OD/NIH HHS/United States
- OT2 OD026553/OD/NIH HHS/United States
- OT2 OD025276/OD/NIH HHS/United States
- OT2 OD026554/OD/NIH HHS/United States
- U24 OD023163/OD/NIH HHS/United States
- T32 HL007604/HL/NHLBI NIH HHS/United States
- OT2 OD023206/OD/NIH HHS/United States
- OT2 OD026556/OD/NIH HHS/United States
- R00 HG012223/HG/NHGRI NIH HHS/United States
- U24 OD023176/OD/NIH HHS/United States
- OT2 OD026548/OD/NIH HHS/United States
- R01 HG010480/HG/NHGRI NIH HHS/United States
- U19 CA203654/CA/NCI NIH HHS/United States
- OT2 OD025315/OD/NIH HHS/United States
- U01 CA249866/CA/NCI NIH HHS/United States
- K99 CA256513/CA/NCI NIH HHS/United States
- OT2 OD026552/OD/NIH HHS/United States
- R01 HL163560/HL/NHLBI NIH HHS/United States
- OT2 OD023205/OD/NIH HHS/United States
- K99 HG012956/HG/NHGRI NIH HHS/United States
- OT2 OD026557/OD/NIH HHS/United States
- U01 HG009088/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
