

Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens
- PMID: 37749268
- PMCID: PMC10961254
- DOI: 10.1038/s41587-023-01948-9
Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens
Abstract
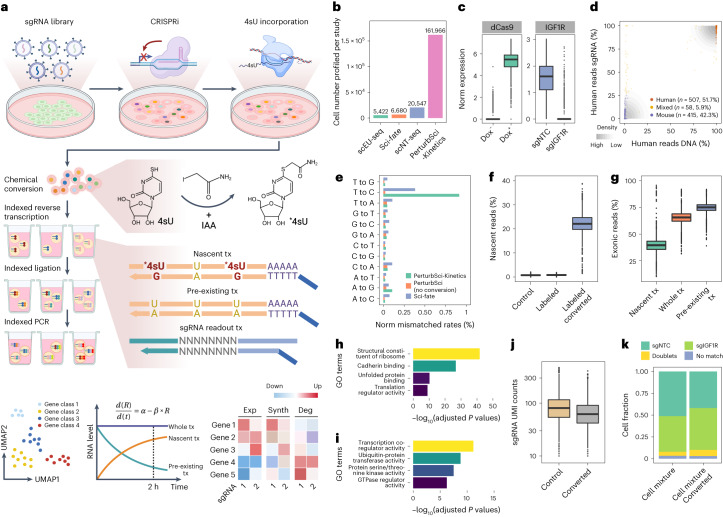
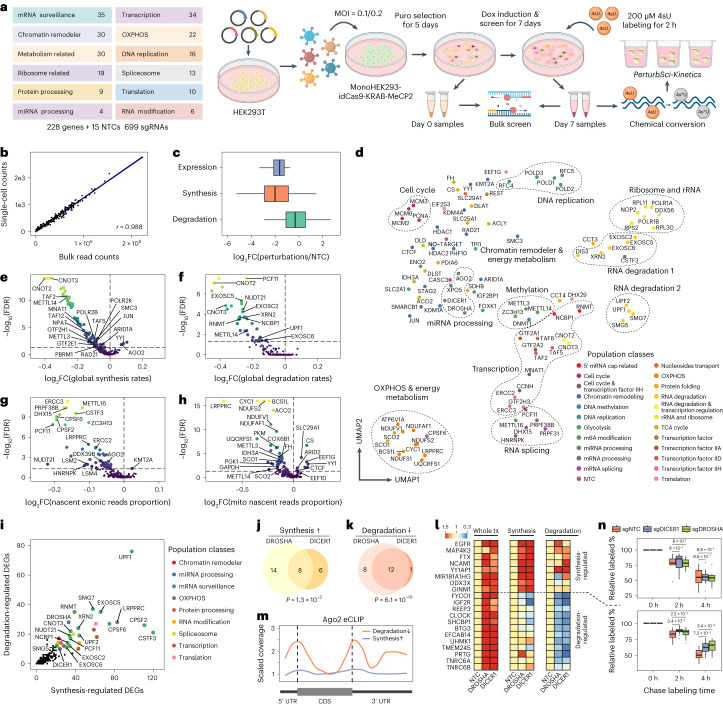
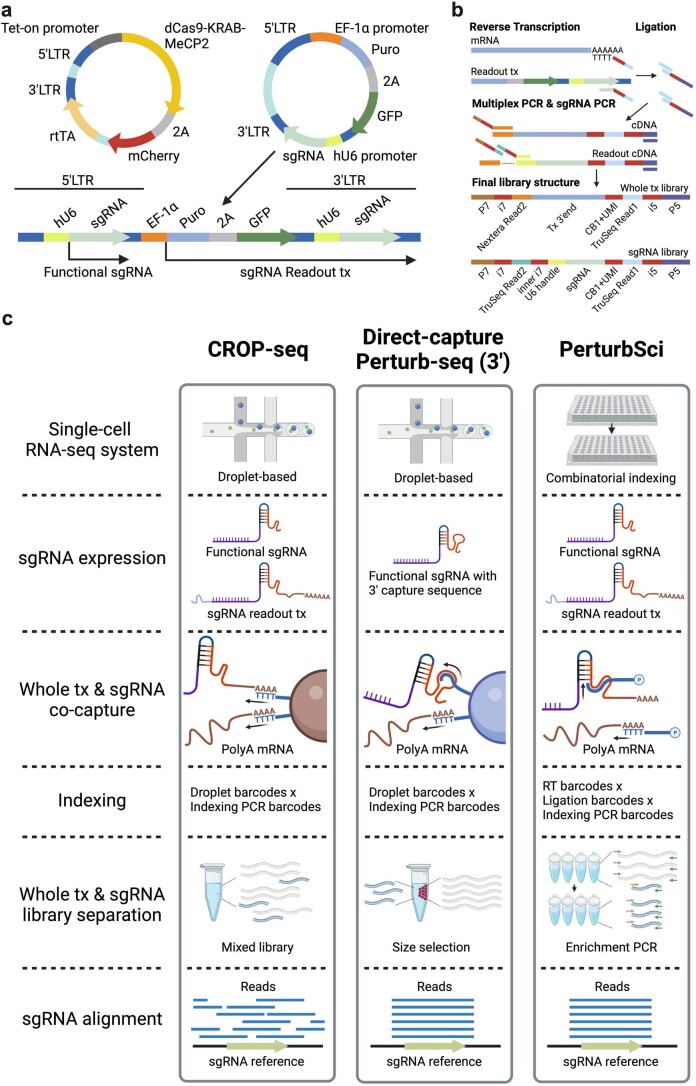
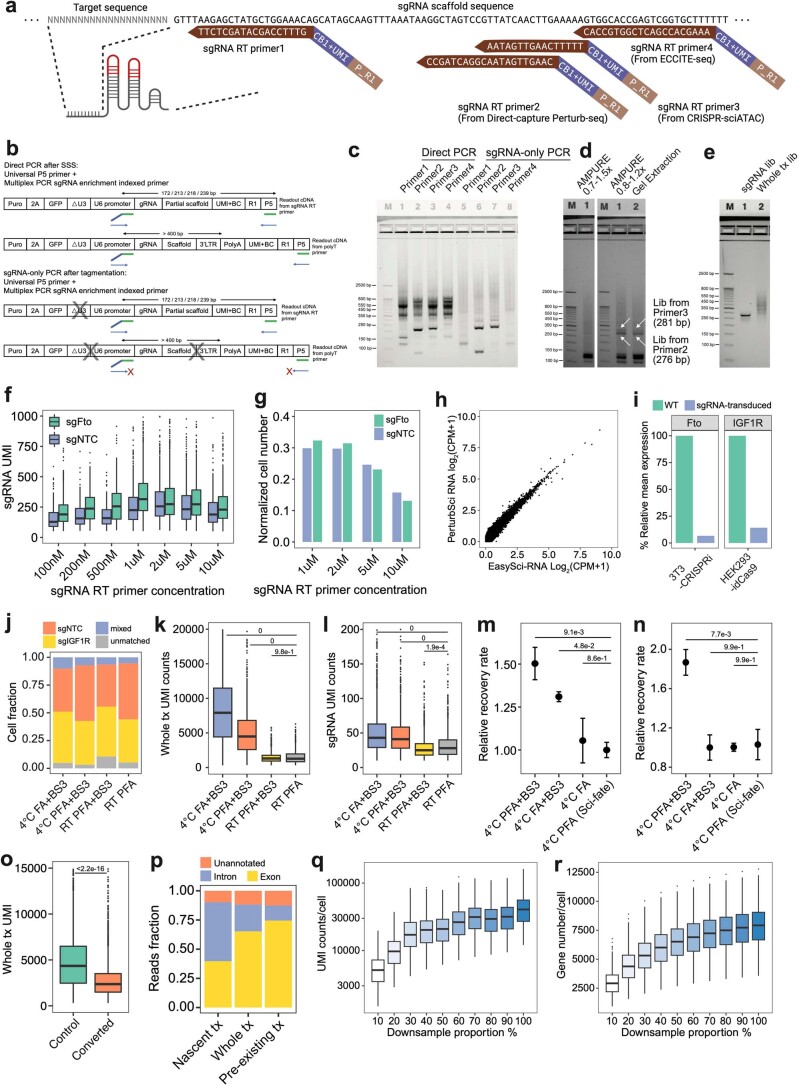
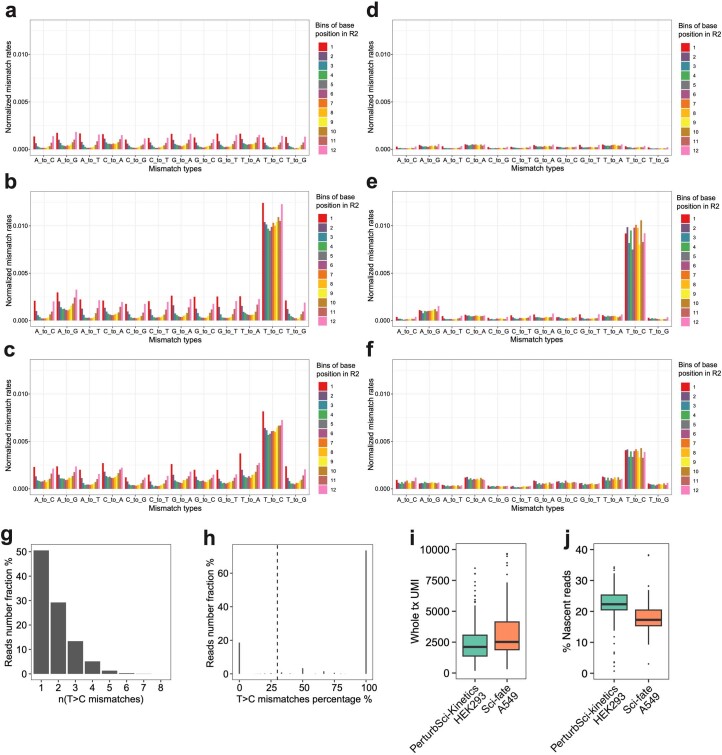
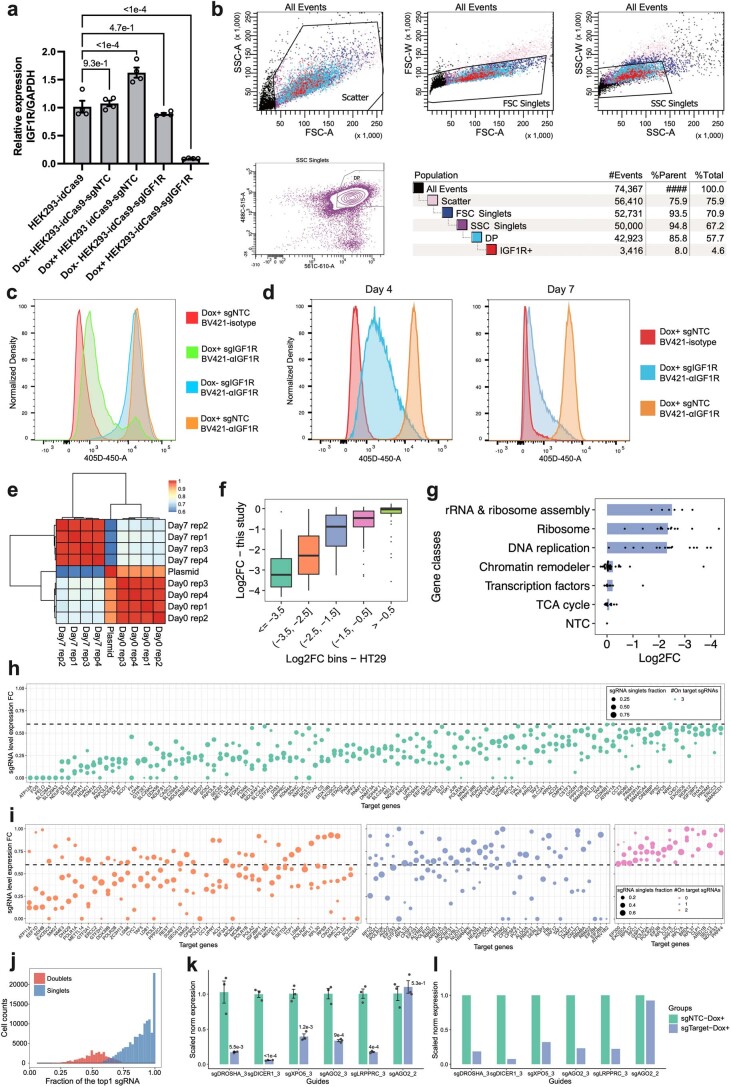
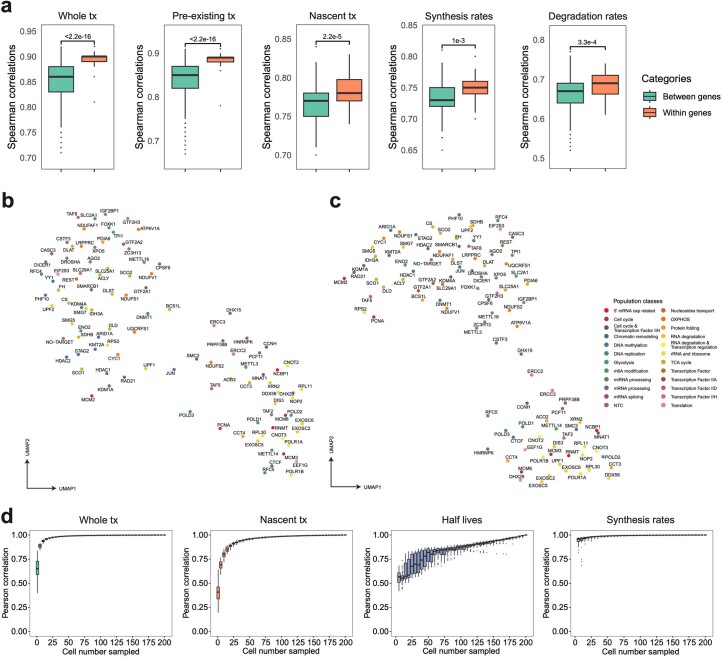
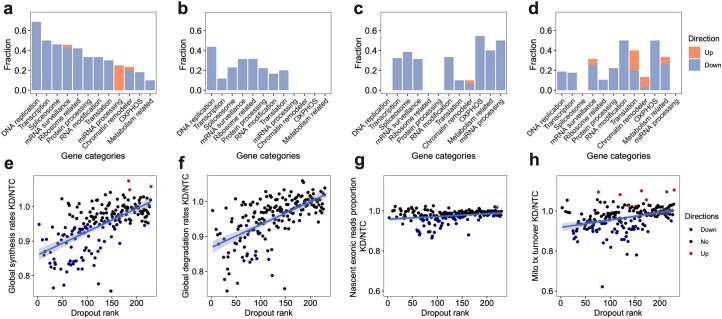
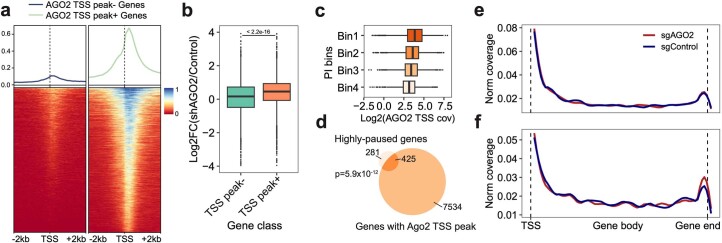
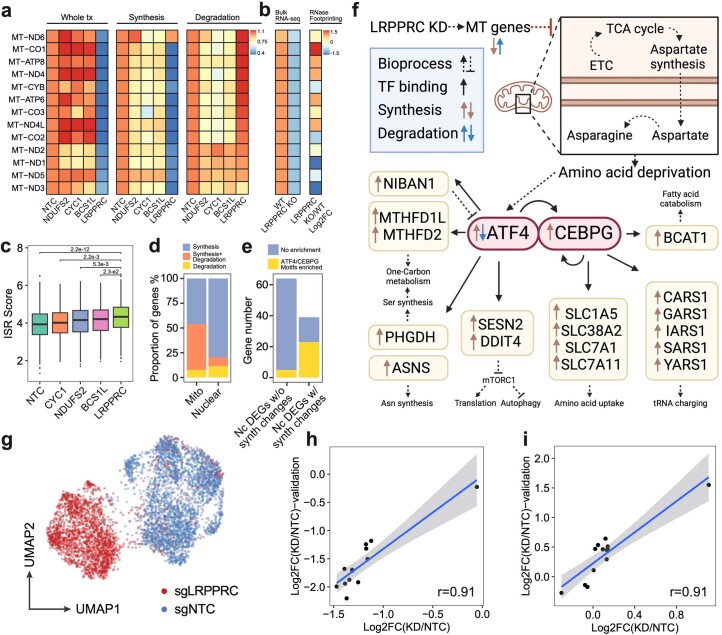
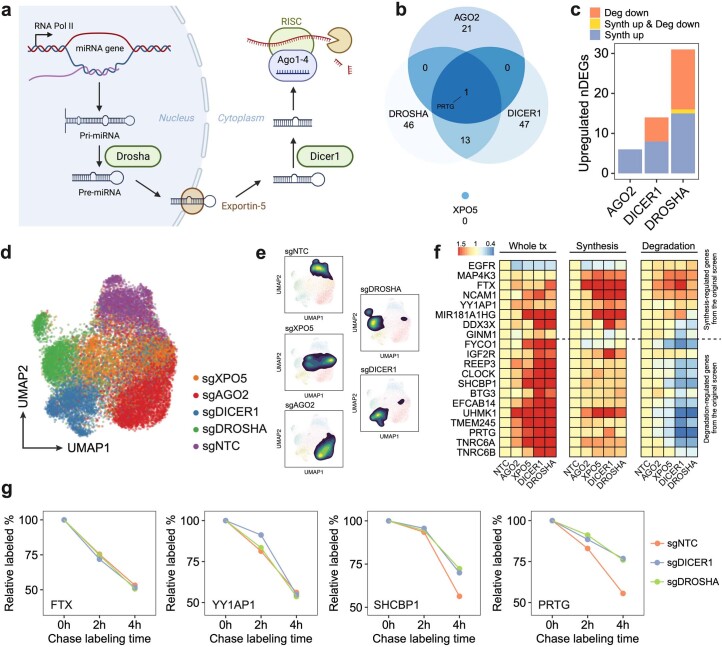
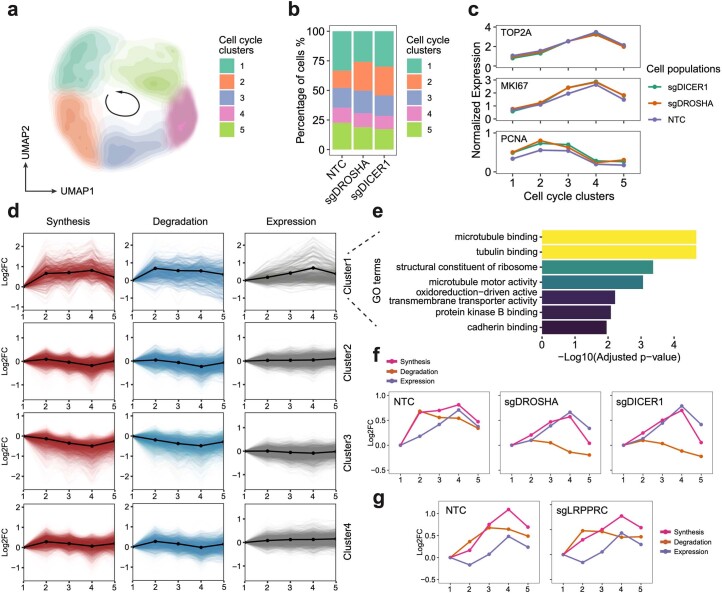
We present a combinatorial indexing method, PerturbSci-Kinetics, for capturing whole transcriptomes, nascent transcriptomes and single guide RNA (sgRNA) identities across hundreds of genetic perturbations at the single-cell level. Profiling a pooled CRISPR screen targeting various biological processes, we show the gene expression regulation during RNA synthesis, processing and degradation, miRNA biogenesis and mitochondrial mRNA processing, systematically decoding the genome-wide regulatory network that underlies RNA temporal dynamics at scale.
© 2023. The Author(s).
Conflict of interest statement
J.C., W.Z. and Z.X. are listed as inventors on a patent related to
Figures
Update of
-
PerturbSci-Kinetics: Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens.bioRxiv [Preprint]. 2023 May 11:2023.01.29.526143. doi: 10.1101/2023.01.29.526143. bioRxiv. 2023. Update in: Nat Biotechnol. 2024 Aug;42(8):1218-1223. doi: 10.1038/s41587-023-01948-9. PMID: 36778497 Free PMC article. Updated. Preprint.
Similar articles
-
PerturbSci-Kinetics: Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens.bioRxiv [Preprint]. 2023 May 11:2023.01.29.526143. doi: 10.1101/2023.01.29.526143. bioRxiv. 2023. Update in: Nat Biotechnol. 2024 Aug;42(8):1218-1223. doi: 10.1038/s41587-023-01948-9. PMID: 36778497 Free PMC article. Updated. Preprint.
-
Pooled CRISPR screening with single-cell transcriptome readout.Nat Methods. 2017 Mar;14(3):297-301. doi: 10.1038/nmeth.4177. Epub 2017 Jan 18. Nat Methods. 2017. PMID: 28099430 Free PMC article.
-
Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells.Nat Methods. 2019 May;16(5):409-412. doi: 10.1038/s41592-019-0392-0. Epub 2019 Apr 22. Nat Methods. 2019. PMID: 31011186 Free PMC article.
-
CRISPR-Based Technologies: Impact of RNA-Targeting Systems.Mol Cell. 2018 Nov 1;72(3):404-412. doi: 10.1016/j.molcel.2018.09.018. Mol Cell. 2018. PMID: 30388409 Free PMC article. Review.
-
Functional Genomics via CRISPR-Cas.J Mol Biol. 2019 Jan 4;431(1):48-65. doi: 10.1016/j.jmb.2018.06.034. Epub 2018 Jun 28. J Mol Biol. 2019. PMID: 29959923 Free PMC article. Review.
Cited by
-
Next-generation forward genetic screens: uniting high-throughput perturbations with single-cell analysis.Trends Genet. 2024 Feb;40(2):118-133. doi: 10.1016/j.tig.2023.10.012. Epub 2023 Nov 20. Trends Genet. 2024. PMID: 37989654 Free PMC article. Review.
-
Multiplex, single-cell CRISPRa screening for cell type specific regulatory elements.Nat Commun. 2024 Sep 18;15(1):8209. doi: 10.1038/s41467-024-52490-4. Nat Commun. 2024. PMID: 39294132 Free PMC article.
-
Transcriptome and Temporal Transcriptome Analyses in Single Cells.Int J Mol Sci. 2024 Nov 29;25(23):12845. doi: 10.3390/ijms252312845. Int J Mol Sci. 2024. PMID: 39684556 Free PMC article. Review.
-
Transcriptome-wide characterization of genetic perturbations.bioRxiv [Preprint]. 2024 Jul 3:2024.07.03.601903. doi: 10.1101/2024.07.03.601903. bioRxiv. 2024. Update in: Nat Genet. 2025 May;57(5):1228-1237. doi: 10.1038/s41588-025-02169-3. PMID: 39005298 Free PMC article. Updated. Preprint.
-
DNA nanodevices map intracellular ions.Nat Biotechnol. 2024 Jul;42(7):1030-1032. doi: 10.1038/s41587-023-02054-6. Nat Biotechnol. 2024. PMID: 38012452 No abstract available.
References
MeSH terms
Substances
Grants and funding
- DP2 HG012522/HG/NHGRI NIH HHS/United States
- R01 AG076932/AG/NIA NIH HHS/United States
- RM1 HG011014/HG/NHGRI NIH HHS/United States
- 1DP2HG012522/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- RM1HG011014/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
