

Single-cell lineage capture across genomic modalities with CellTag-multi reveals fate-specific gene regulatory changes
- PMID: 37749269
- PMCID: PMC11180607
- DOI: 10.1038/s41587-023-01931-4
Single-cell lineage capture across genomic modalities with CellTag-multi reveals fate-specific gene regulatory changes
Abstract
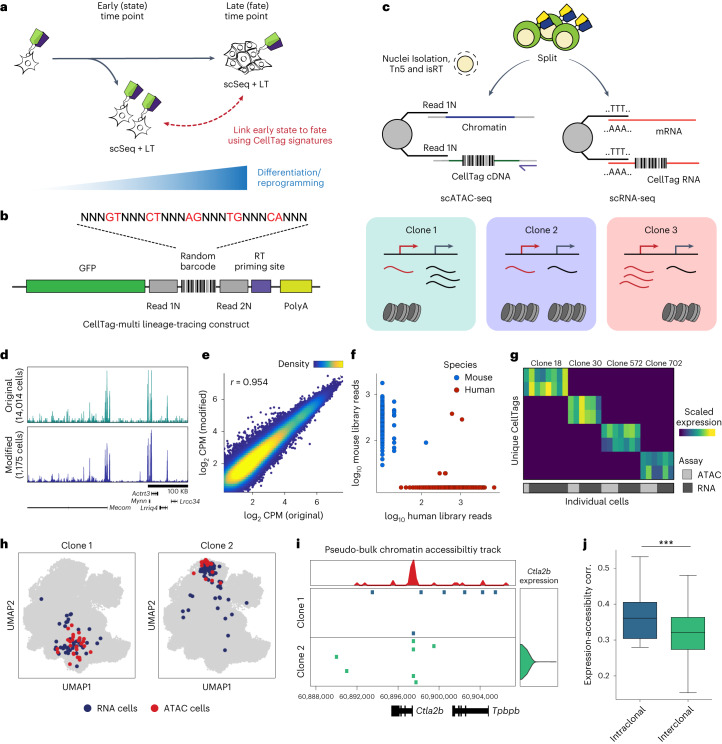
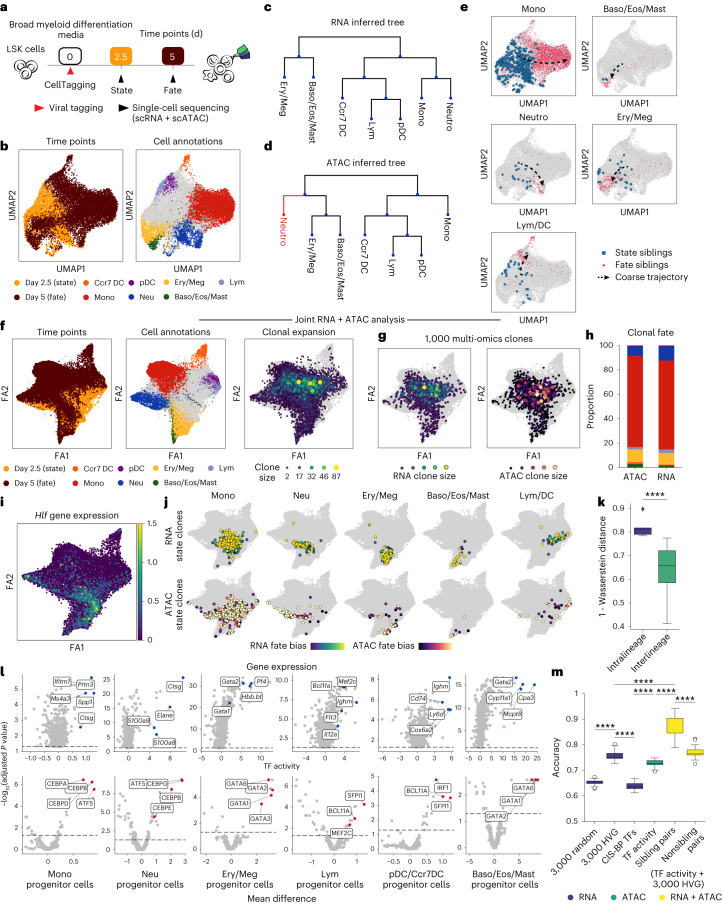
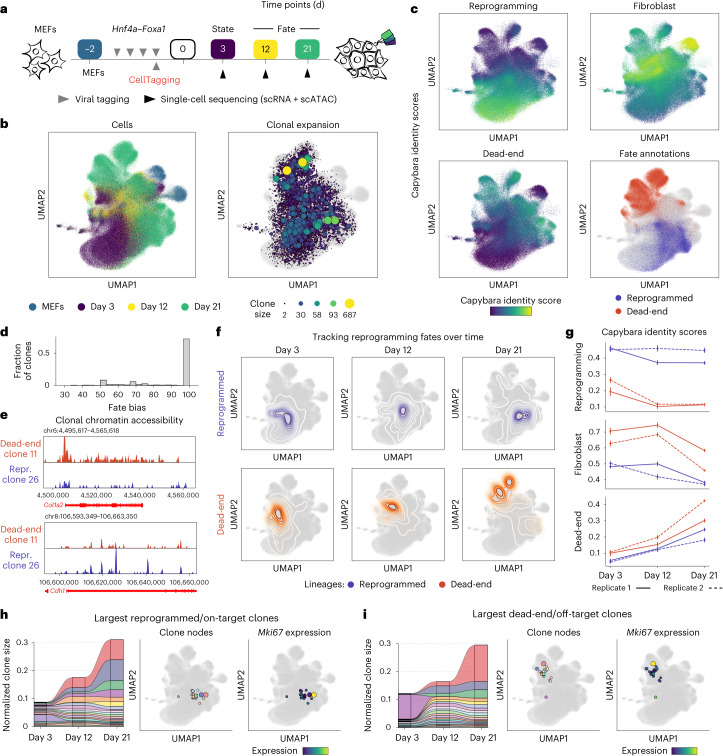
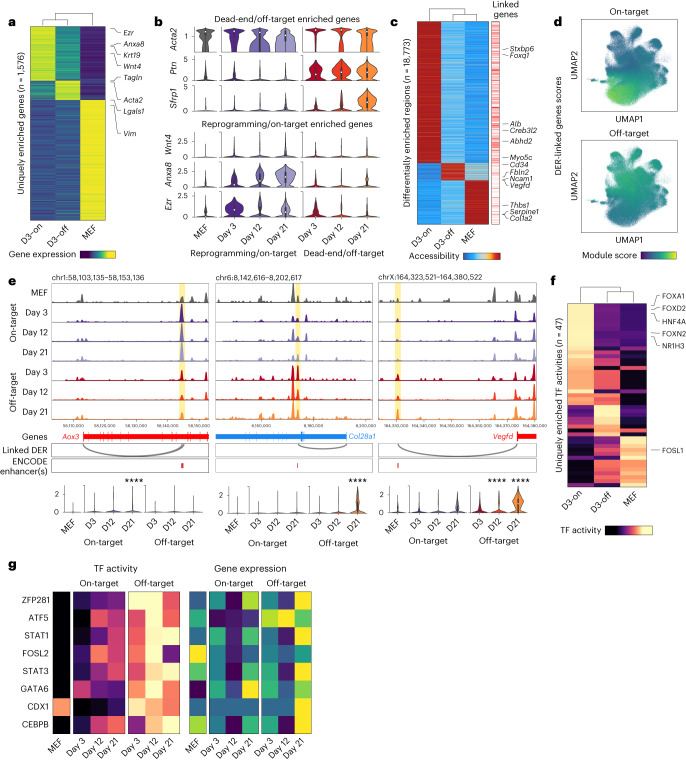
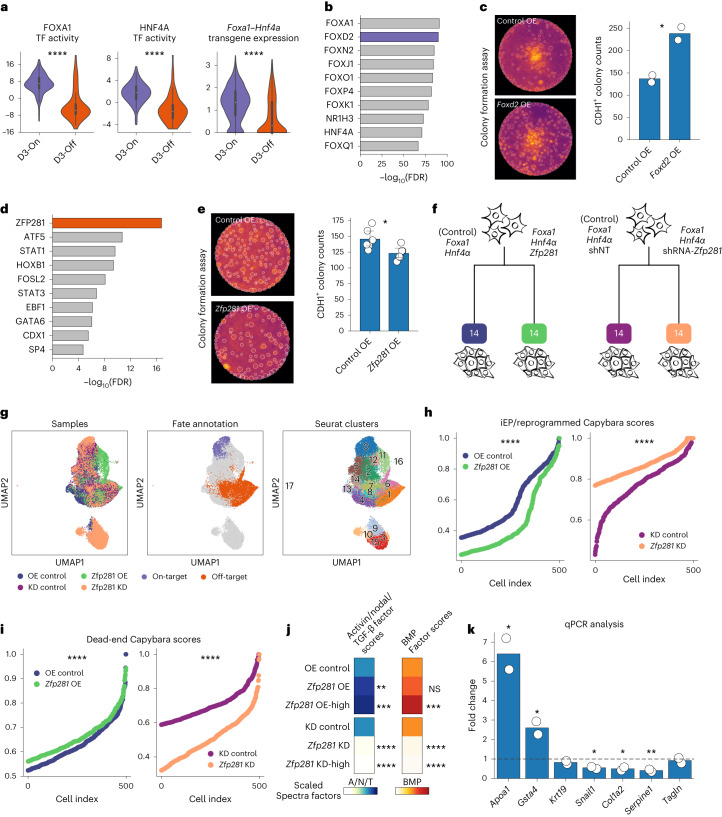
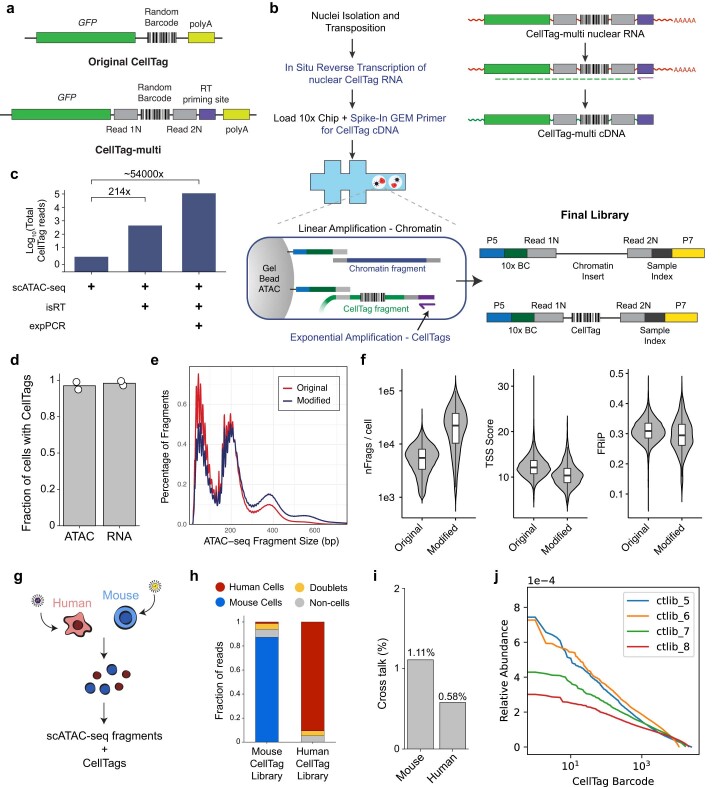
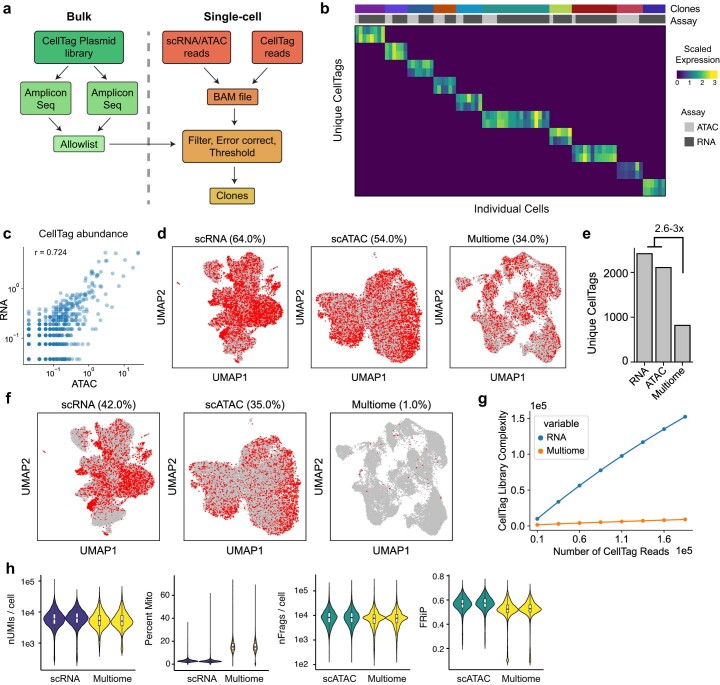
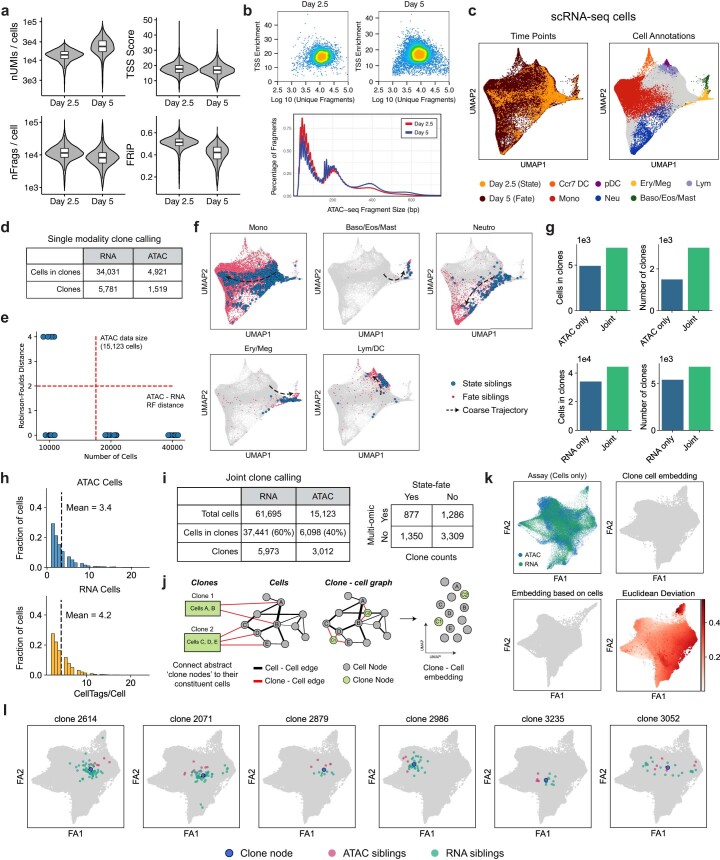
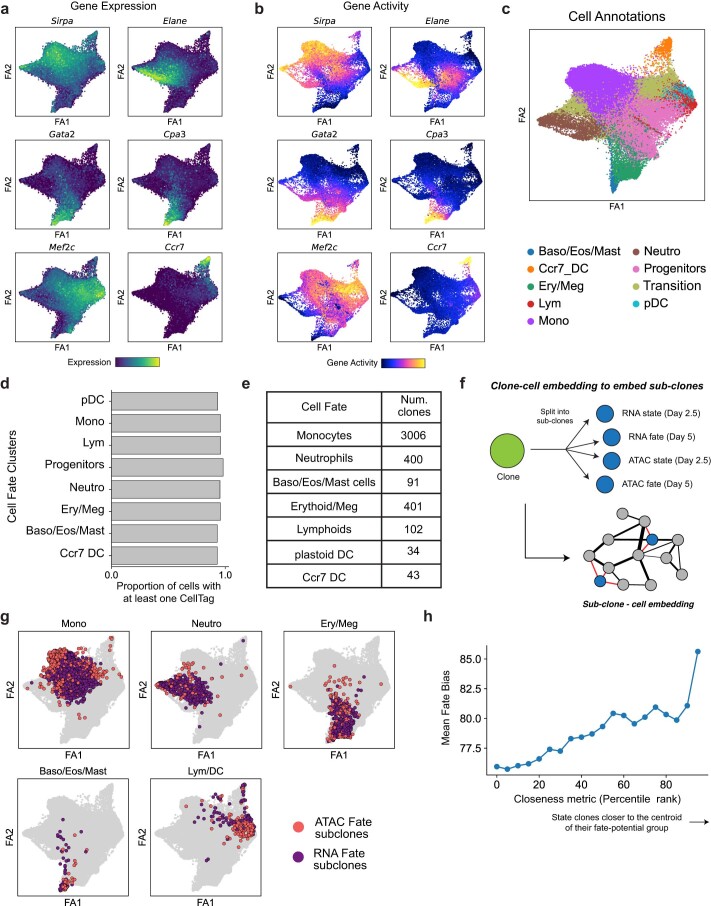
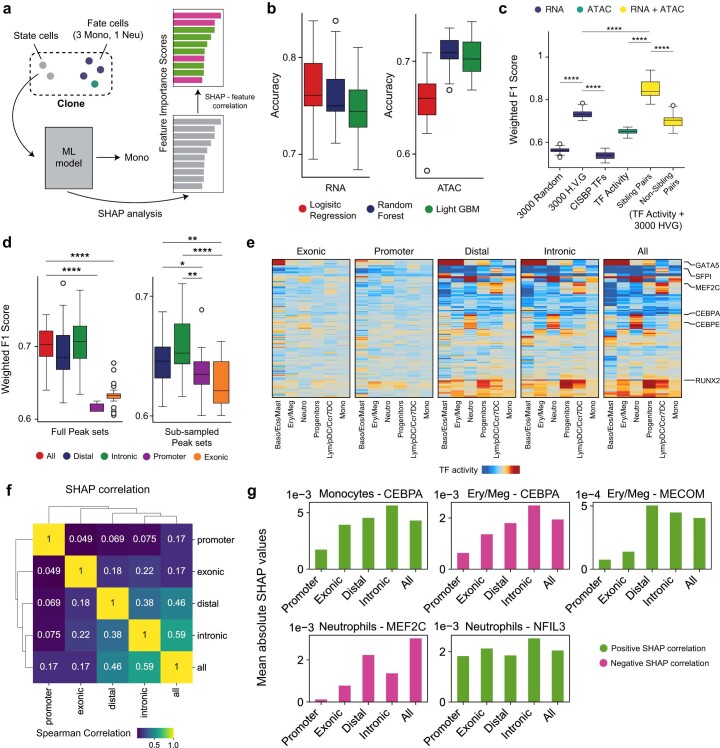
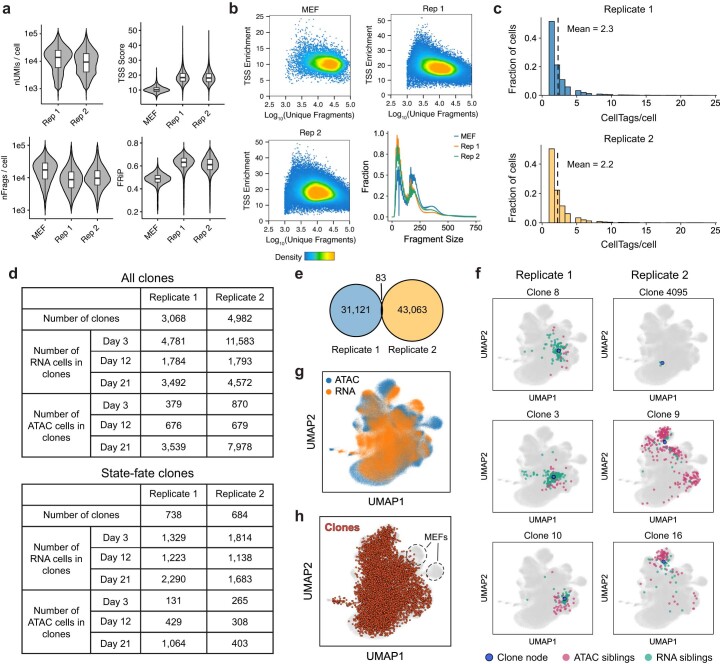
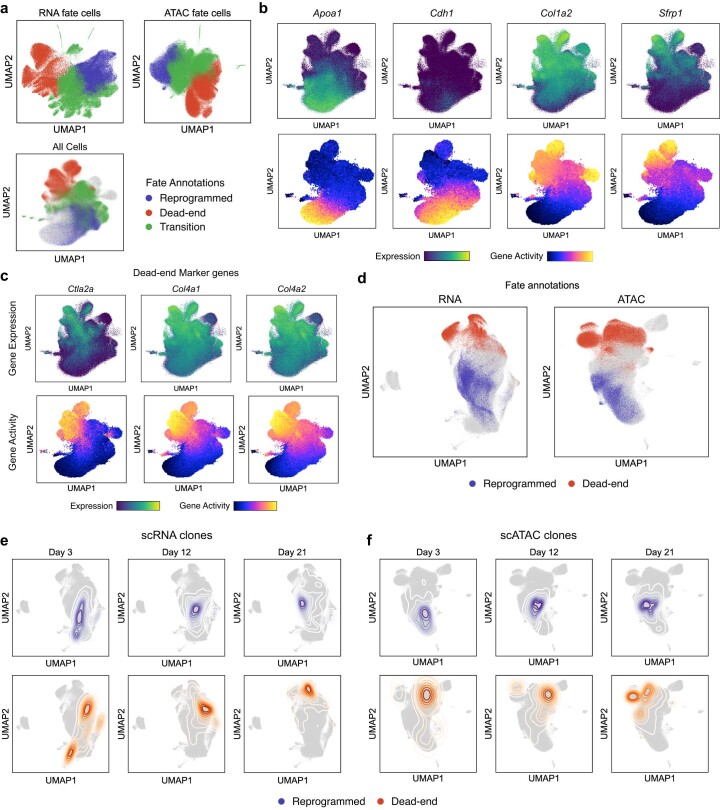
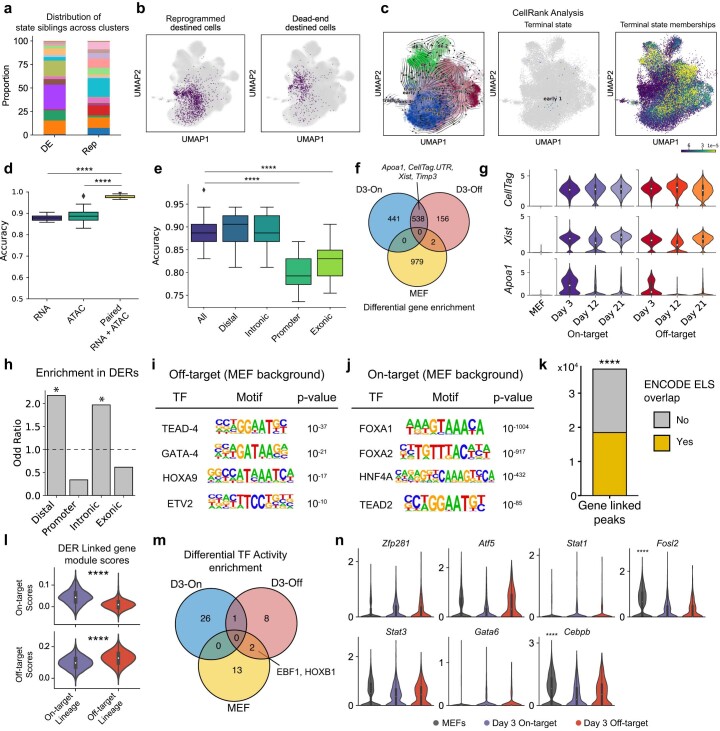
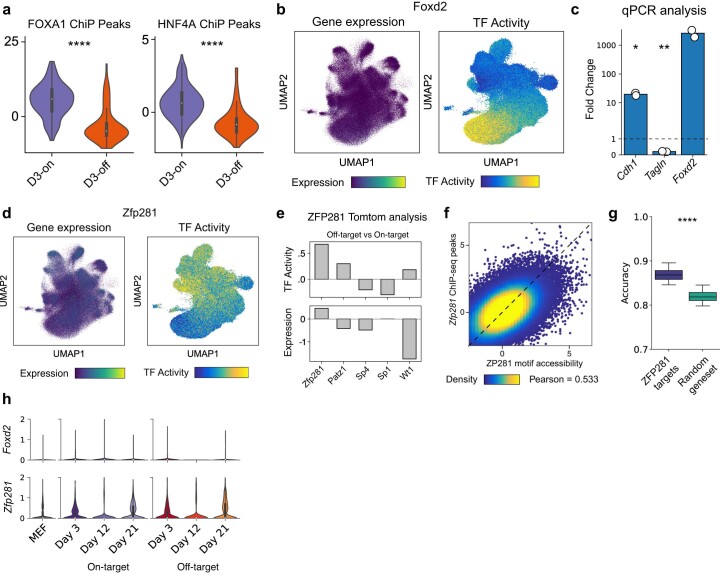
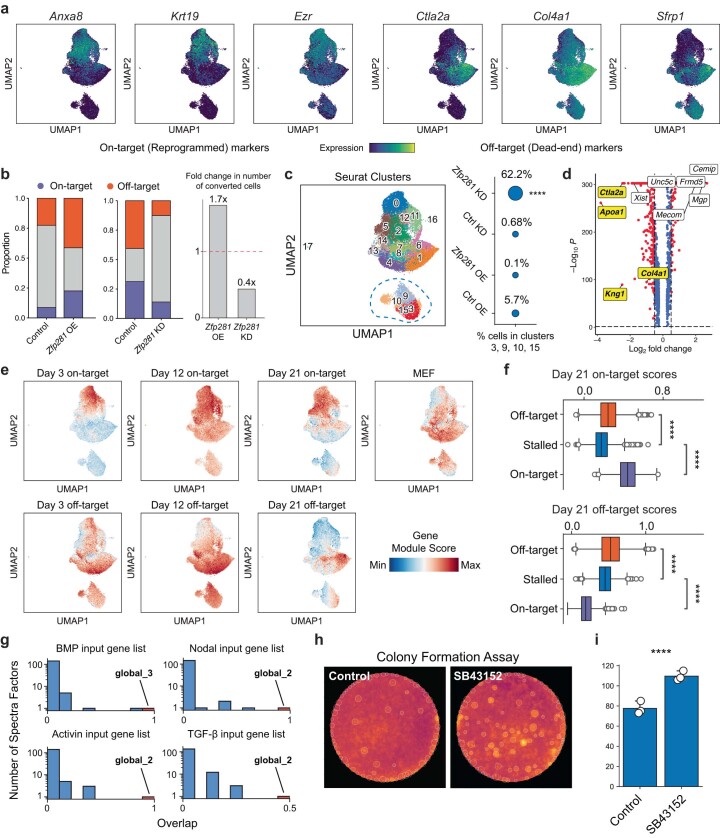
Complex gene regulatory mechanisms underlie differentiation and reprogramming. Contemporary single-cell lineage-tracing (scLT) methods use expressed, heritable DNA barcodes to combine cell lineage readout with single-cell transcriptomics. However, reliance on transcriptional profiling limits adaptation to other single-cell assays. With CellTag-multi, we present an approach that enables direct capture of heritable random barcodes expressed as polyadenylated transcripts, in both single-cell RNA sequencing and single-cell Assay for Transposase Accessible Chromatin using sequencing assays, allowing for independent clonal tracking of transcriptional and epigenomic cell states. We validate CellTag-multi to characterize progenitor cell lineage priming during mouse hematopoiesis. Additionally, in direct reprogramming of fibroblasts to endoderm progenitors, we identify core regulatory programs underlying on-target and off-target fates. Furthermore, we reveal the transcription factor Zfp281 as a regulator of reprogramming outcome, biasing cells toward an off-target mesenchymal fate. Our results establish CellTag-multi as a lineage-tracing method compatible with multiple single-cell modalities and demonstrate its utility in revealing fate-specifying gene regulatory changes across diverse paradigms of differentiation and reprogramming.
© 2023. The Author(s).
Conflict of interest statement
S.A.M. and K.J. are named inventors on a patent application for this technology. S.A.M. and G.R.G. are cofounders of CapyBio. The remaining authors declare no conflict of interest.
Figures
References
-
- Clevers H, et al. What is your conceptual definition of ‘cell type’ in the context of a mature organism? Cell Syst. 2017;4:255–259. - PubMed
-
- Stuart T, Satija R. Integrative single-cell analysis. Nat. Rev. Genet. 2019;20:257–272. - PubMed
-
- Morris SA. The evolving concept of cell identity in the single cell era. Development. 2019;146:dev169748. - PubMed
-
- Kester L, van Oudenaarden A. Single-cell transcriptomics meets lineage tracing. Cell Stem Cell. 2018;23:166–179. - PubMed
-
- VanHorn S, Morris SA. Next-generation lineage tracing and fate mapping to interrogate development. Dev. Cell. 2021;56:7–21. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
