

Rearrangement Events on Circular Genomes
- PMID: 37749280
- PMCID: PMC10520144
- DOI: 10.1007/s11538-023-01209-5
Rearrangement Events on Circular Genomes
Abstract
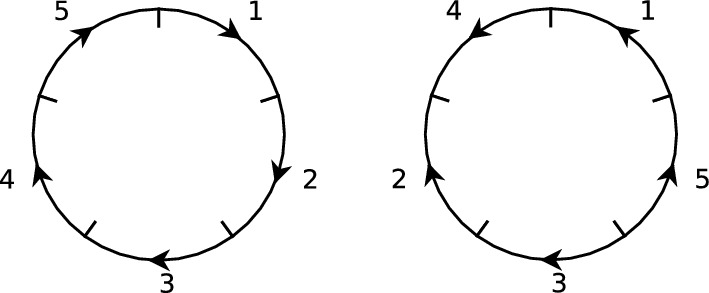
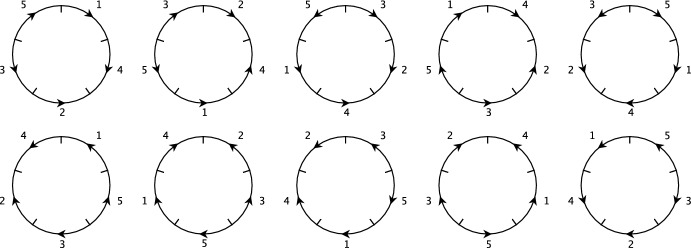
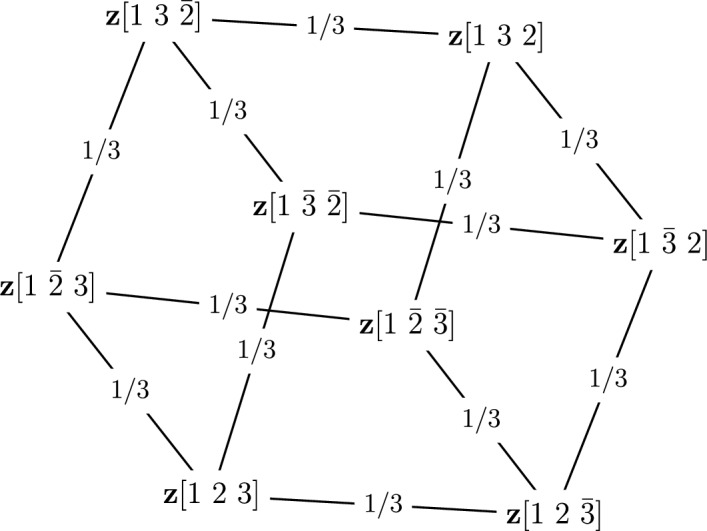
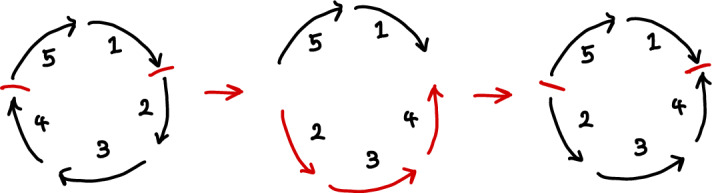
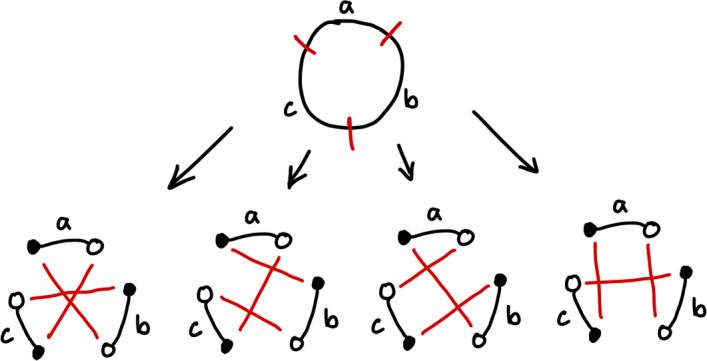
Early literature on genome rearrangement modelling views the problem of computing evolutionary distances as an inherently combinatorial one. In particular, attention is given to estimating distances using the minimum number of events required to transform one genome into another. In hindsight, this approach is analogous to early methods for inferring phylogenetic trees from DNA sequences such as maximum parsimony-both are motivated by the principle that the true distance minimises evolutionary change, and both are effective if this principle is a true reflection of reality. Recent literature considers genome rearrangement under statistical models, continuing this parallel with DNA-based methods, with the goal of using model-based methods (for example maximum likelihood techniques) to compute distance estimates that incorporate the large number of rearrangement paths that can transform one genome into another. Crucially, this approach requires one to decide upon a set of feasible rearrangement events and, in this paper, we focus on characterising well-motivated models for signed, uni-chromosomal circular genomes, where the number of regions remains fixed. Since rearrangements are often mathematically described using permutations, we isolate the sets of permutations representing rearrangements that are biologically reasonable in this context, for example inversions and transpositions. We provide precise mathematical expressions for these rearrangements, and then describe them in terms of the set of cuts made in the genome when they are applied. We directly compare cuts to breakpoints, and use this concept to count the distinct rearrangement actions which apply a given number of cuts. Finally, we provide some examples of rearrangement models, and include a discussion of some questions that arise when defining plausible models.
Keywords: Combinatorics; Genome rearrangements; Phylogenetics.
© 2023. The Author(s).
Figures


References
-
- Alexandrino A, Oliveira A, Dias U, Dias Z. On the complexity of some variations of sorting by transpositions. JUCS J Univ Comput Sci. 2020;26(9):1076–1094. doi: 10.3897/jucs.2020.057. - DOI
-
- Alexeev N, Aidagulov R, Alekseyev MA. A computational method for the rate estimation of evolutionary transpositions. In: Ortuño F, Rojas I, editors. Bioinform Biomed Eng. Cham: Springer; 2015. pp. 471–480.
-
- Bafna V, Pevzner PA. Genome rearrangements and sorting by reversals. SIAM J Comput. 1996;25(2):272–289. doi: 10.1137/s0097539793250627. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
