

RANK ligand converts the NCoR/HDAC3 co-repressor to a PGC1β- and RNA-dependent co-activator of osteoclast gene expression
- PMID: 37751740
- PMCID: PMC10591845
- DOI: 10.1016/j.molcel.2023.08.029
RANK ligand converts the NCoR/HDAC3 co-repressor to a PGC1β- and RNA-dependent co-activator of osteoclast gene expression
Abstract
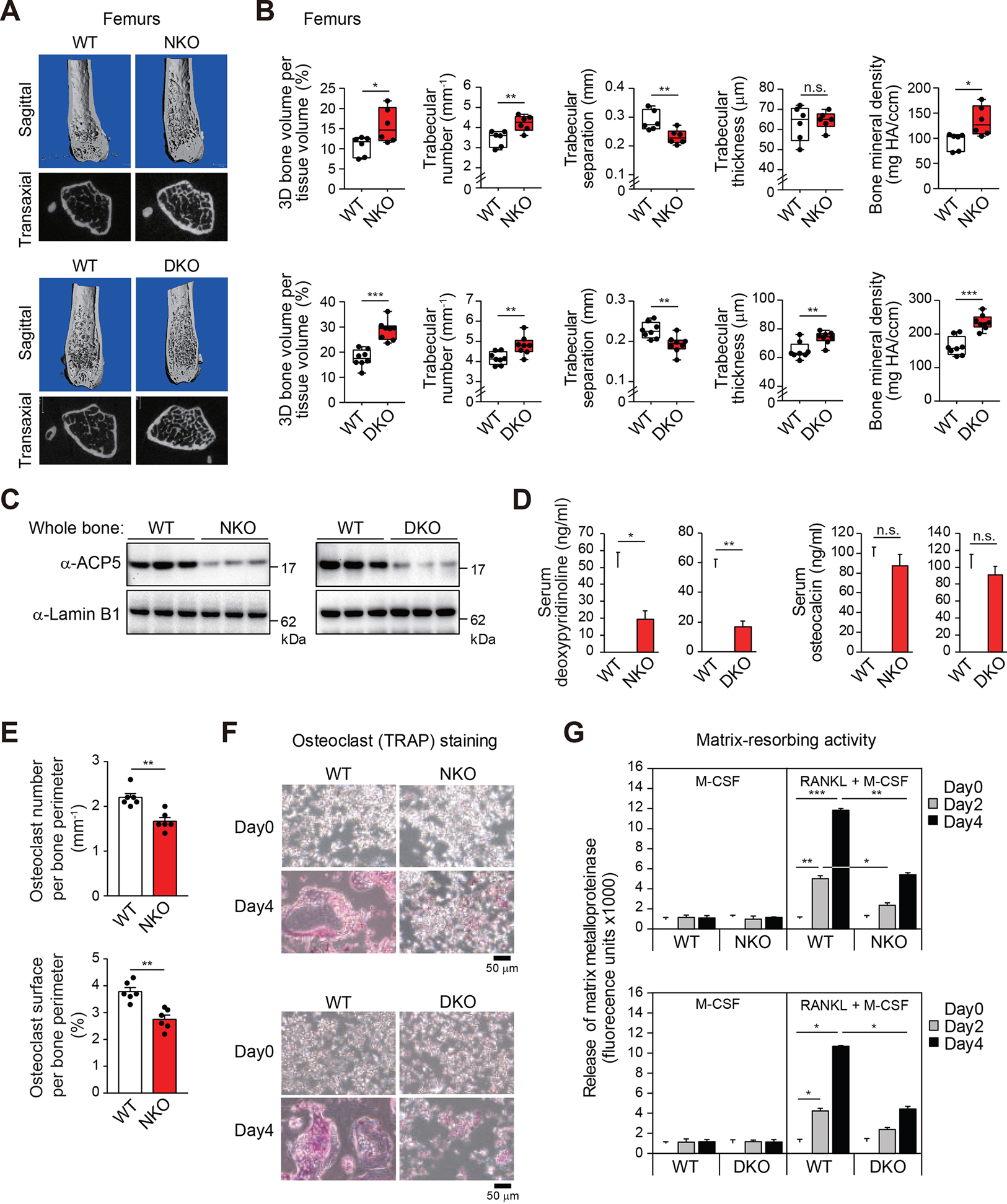
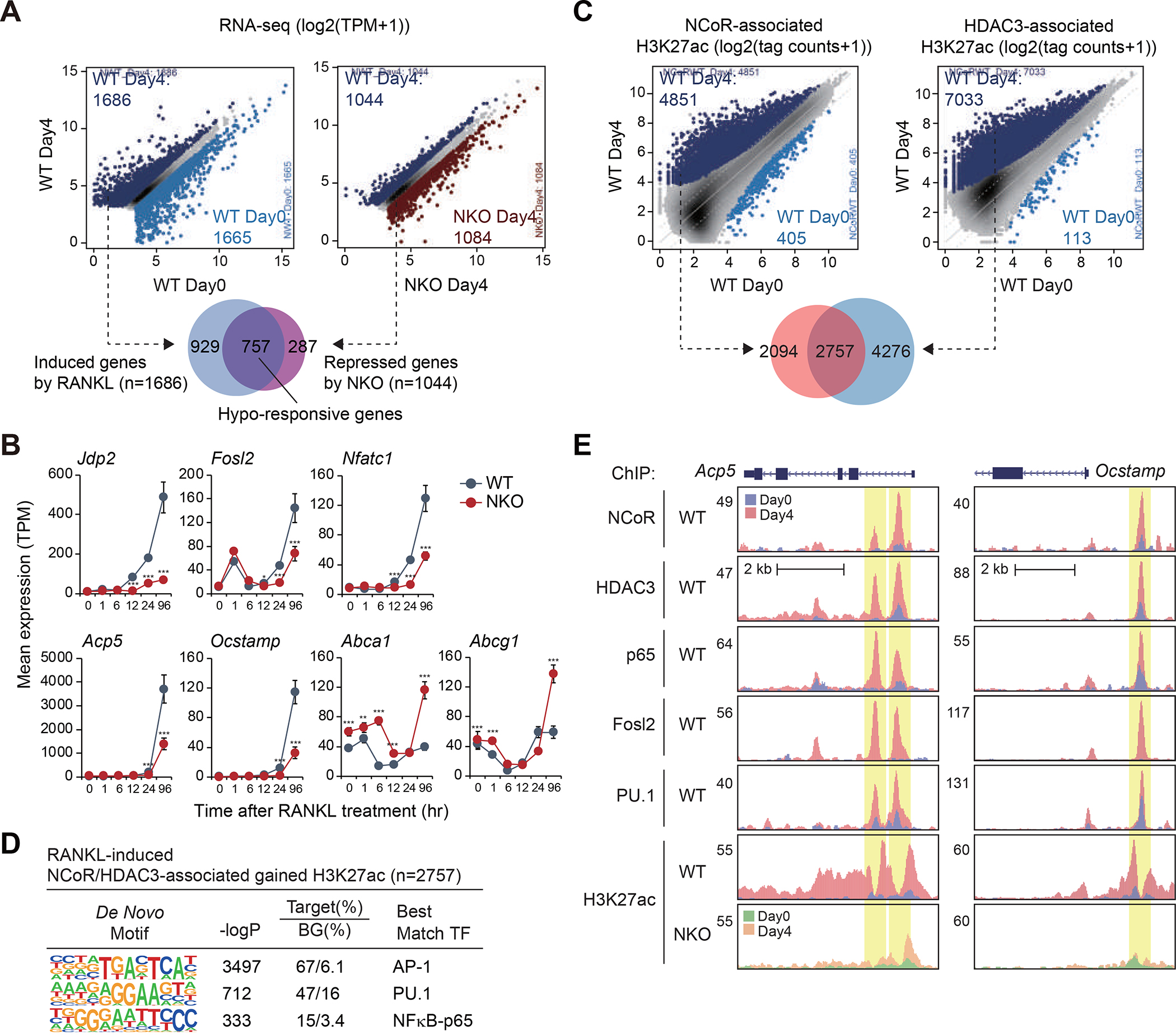
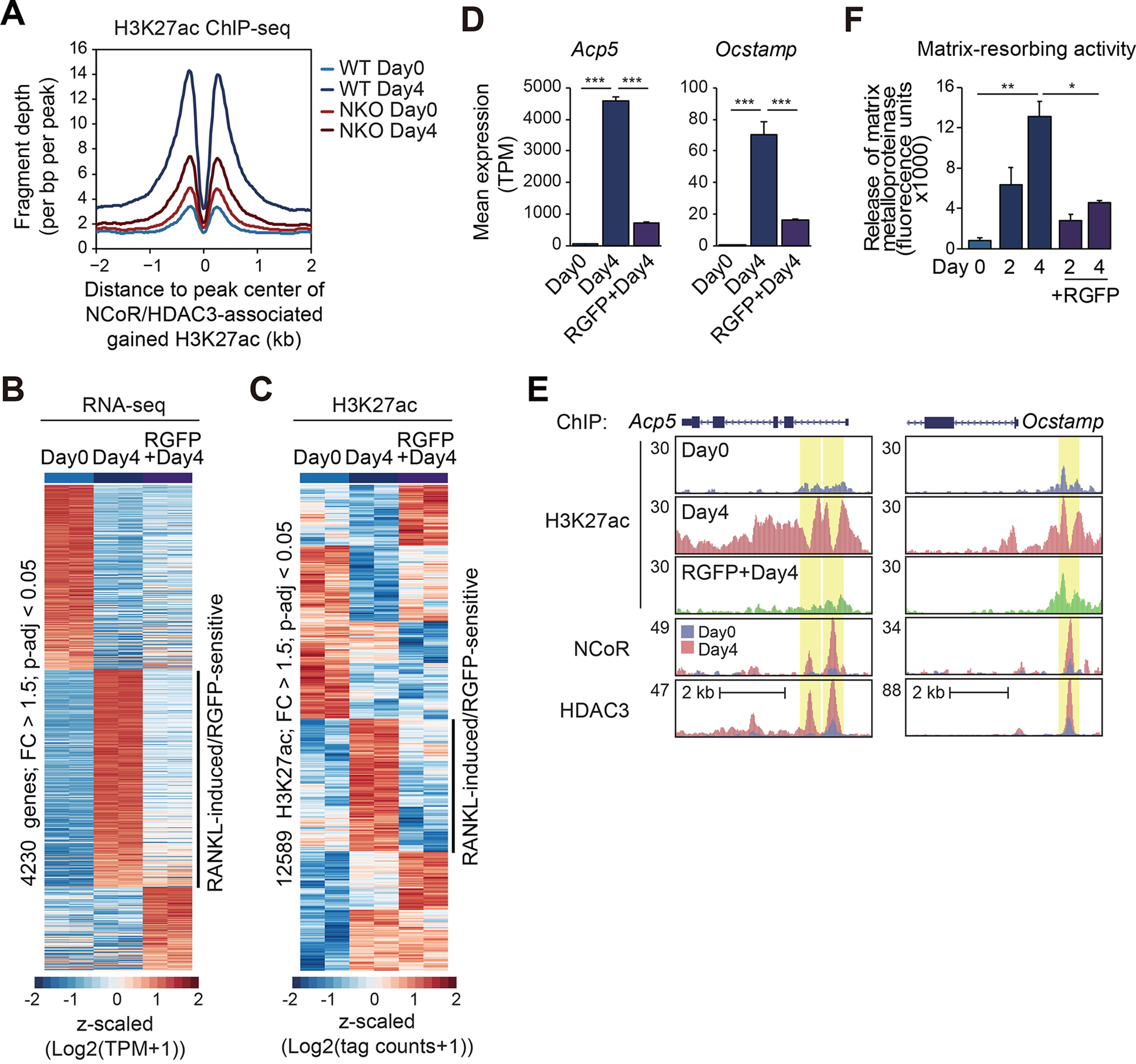
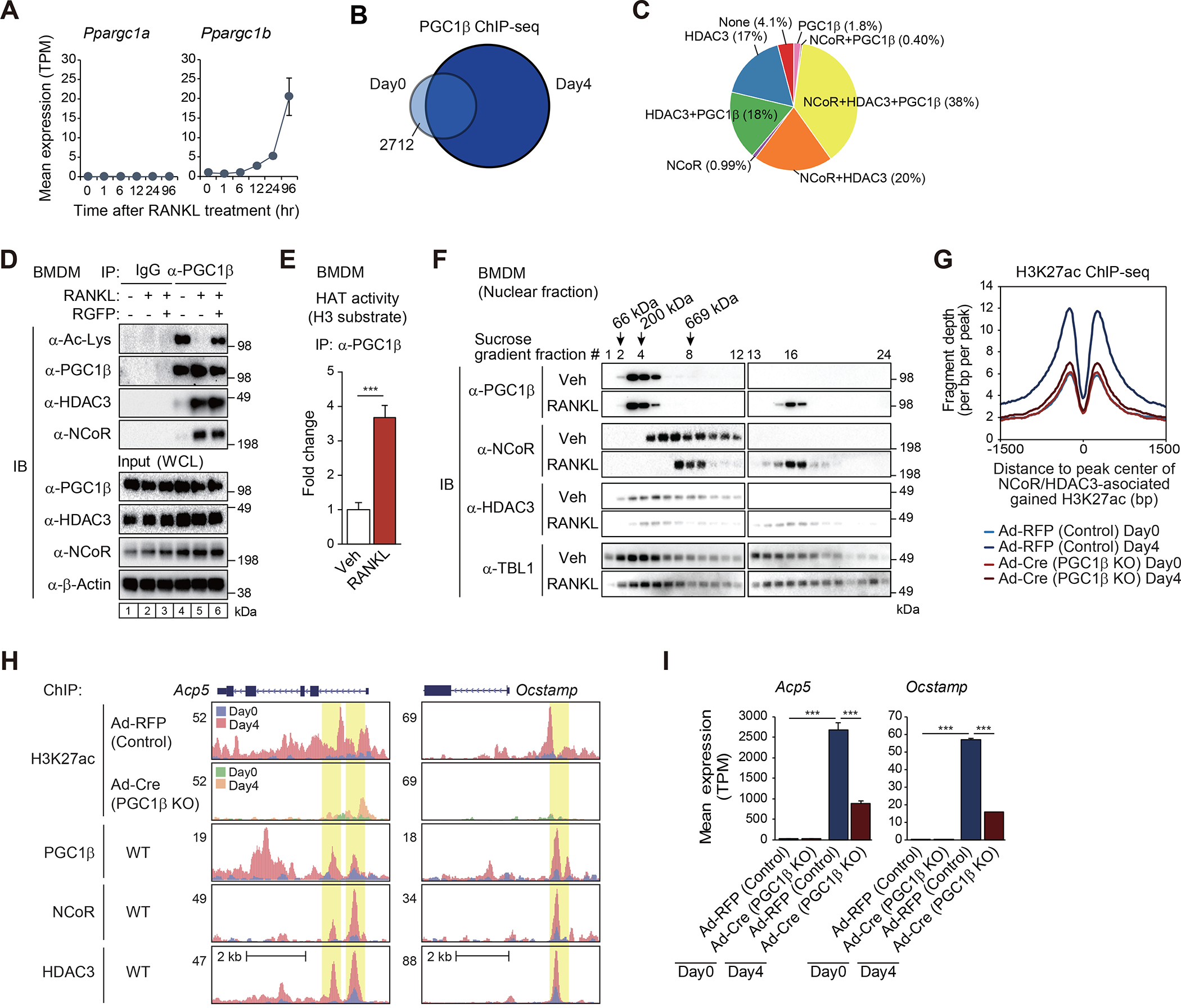
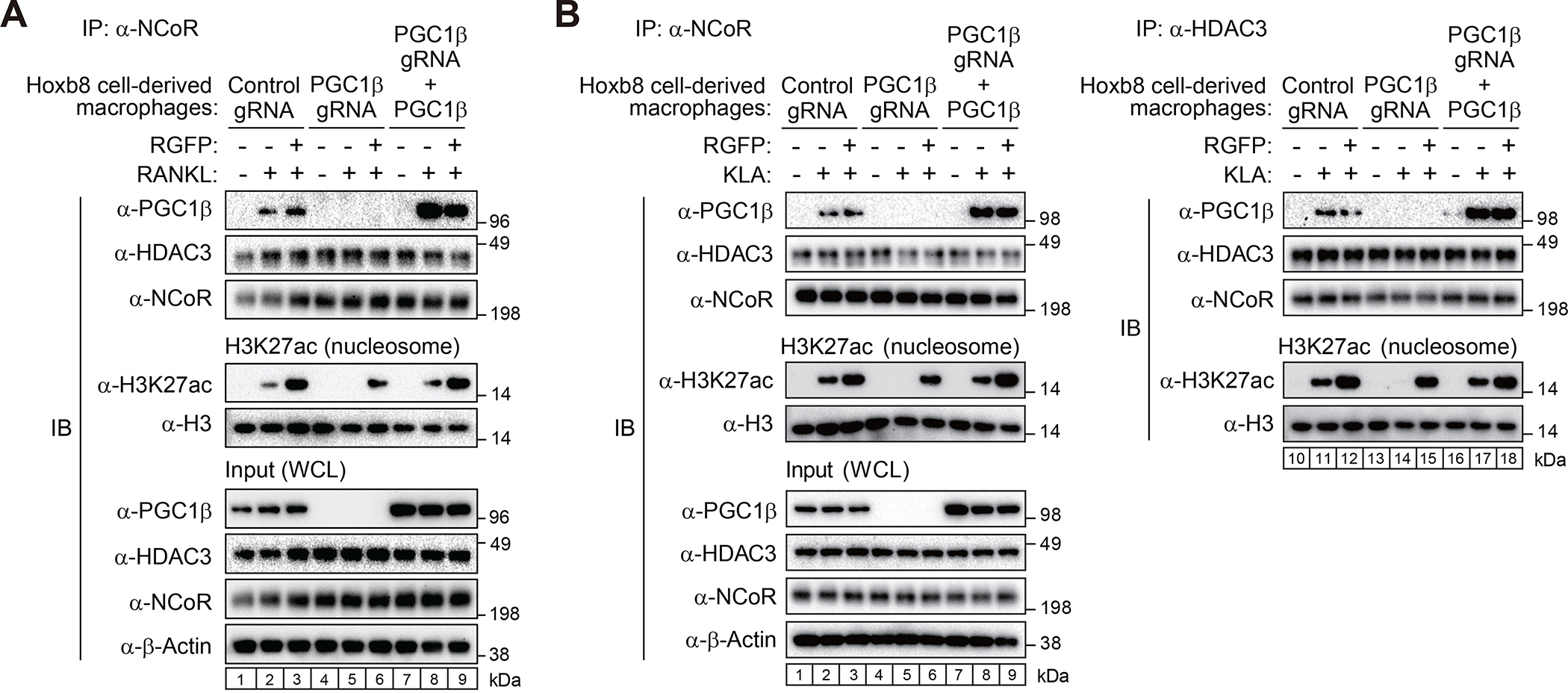
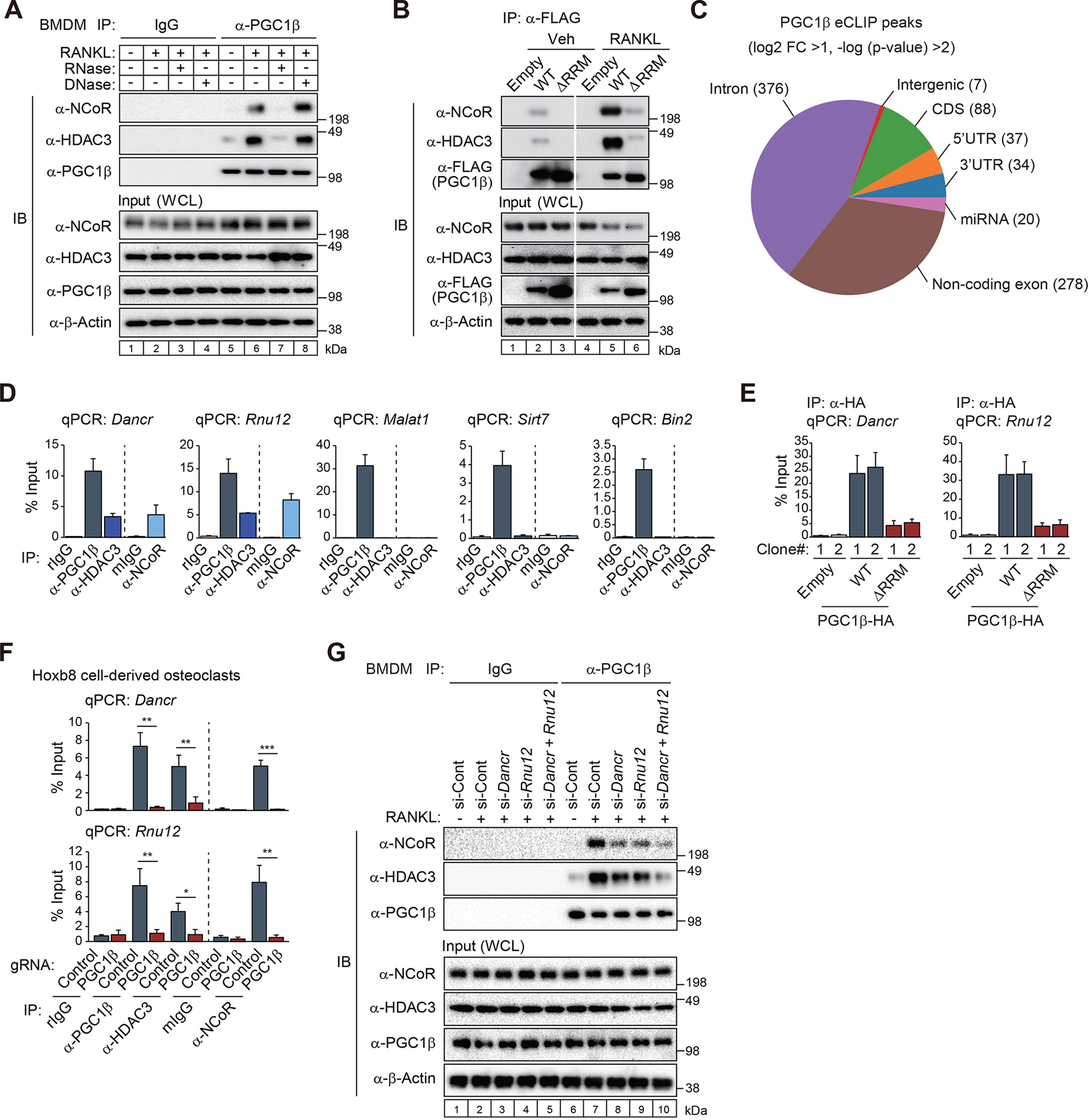
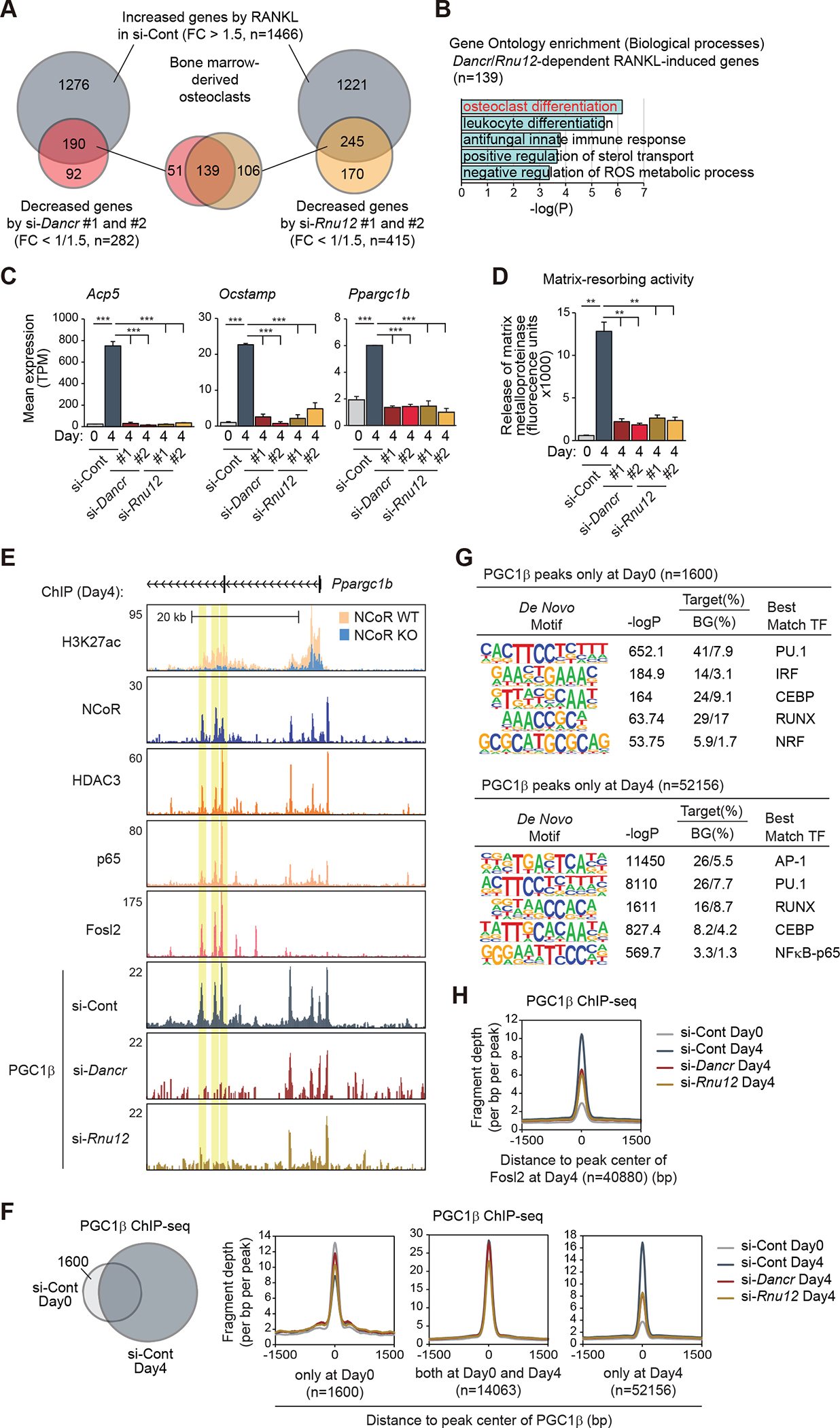
The nuclear receptor co-repressor (NCoR) complex mediates transcriptional repression dependent on histone deacetylation by histone deacetylase 3 (HDAC3) as a component of the complex. Unexpectedly, we found that signaling by the receptor activator of nuclear factor κB (RANK) converts the NCoR/HDAC3 co-repressor complex to a co-activator of AP-1 and NF-κB target genes that are required for mouse osteoclast differentiation. Accordingly, the dominant function of NCoR/HDAC3 complexes in response to RANK signaling is to activate, rather than repress, gene expression. Mechanistically, RANK signaling promotes RNA-dependent interaction of the transcriptional co-activator PGC1β with the NCoR/HDAC3 complex, resulting in the activation of PGC1β and inhibition of HDAC3 activity for acetylated histone H3. Non-coding RNAs Dancr and Rnu12, which are associated with altered human bone homeostasis, promote NCoR/HDAC3 complex assembly and are necessary for RANKL-induced osteoclast differentiation in vitro. These findings may be prototypic for signal-dependent functions of NCoR in other biological contexts.
Keywords: AP-1; H3K27ac; HDAC3; NCoR; NF-κB; PGC1β; RANK; co-activator; deacetylation; gene expression; non-coding RNA; osteoclast.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests C.K.G. is a co-founder, equity holder, and member of the Scientific Advisory Board of Asteroid Therapeutics. J.T.K. is on the Molecular Cell advisory board.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- P01 HL088093/HL/NHLBI NIH HHS/United States
- P20 GM125503/GM/NIGMS NIH HHS/United States
- R35 GM118060/GM/NIGMS NIH HHS/United States
- R01 AG068449/AG/NIA NIH HHS/United States
- R01 DK091183/DK/NIDDK NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- S10 OD026929/OD/NIH HHS/United States
- R01 DK018477/DK/NIDDK NIH HHS/United States
- R01 DK039949/DK/NIDDK NIH HHS/United States
- R21 HL088083/HL/NHLBI NIH HHS/United States
- P01 HL147835/HL/NHLBI NIH HHS/United States
- P50 GM085764/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
