

An organism-wide ATAC-seq peak catalog for the bovine and its use to identify regulatory variants
- PMID: 37751945
- PMCID: PMC10691486
- DOI: 10.1101/gr.277947.123
An organism-wide ATAC-seq peak catalog for the bovine and its use to identify regulatory variants
Abstract
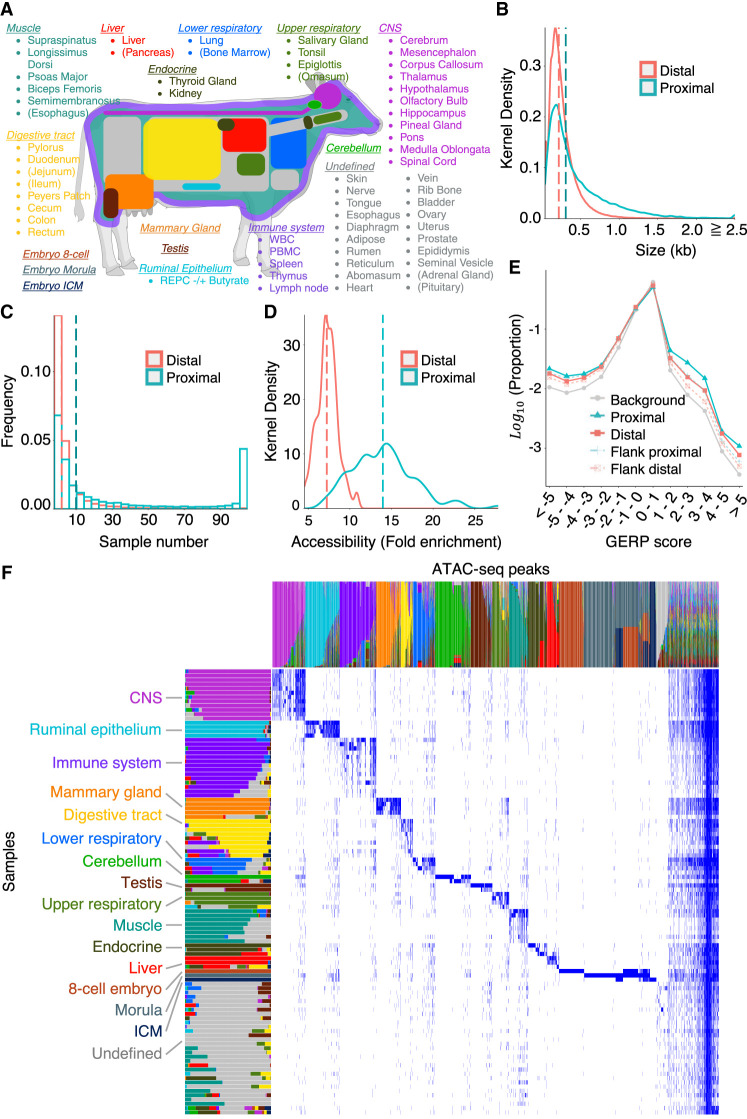
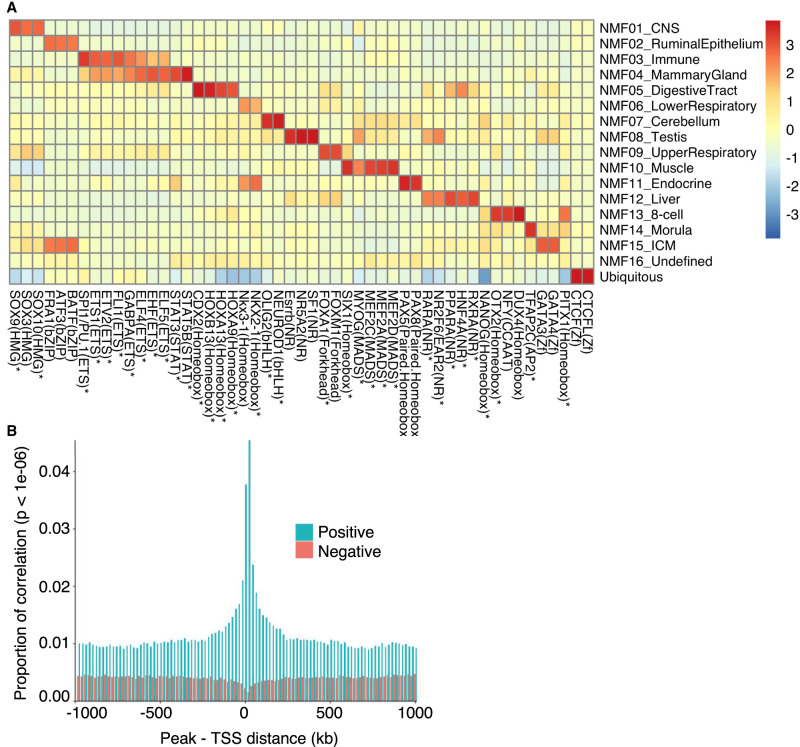
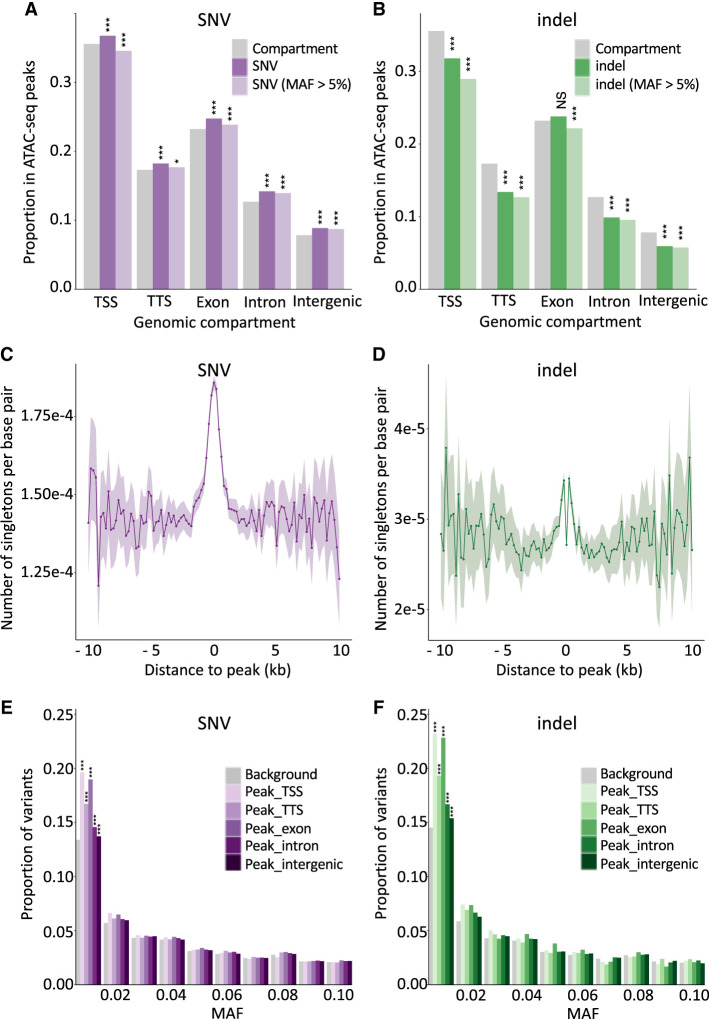
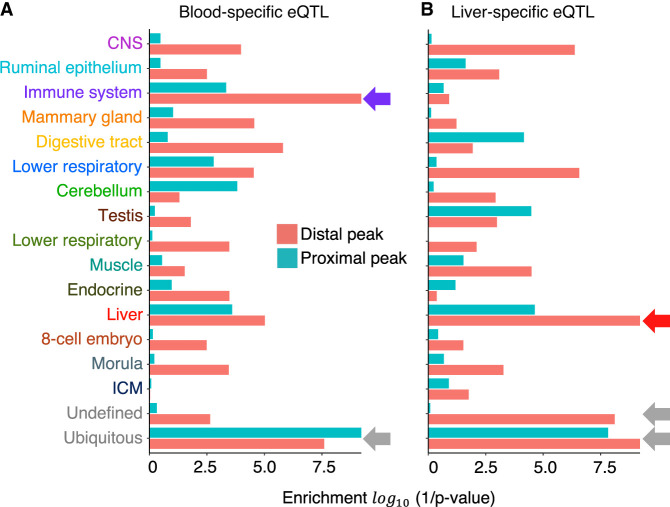
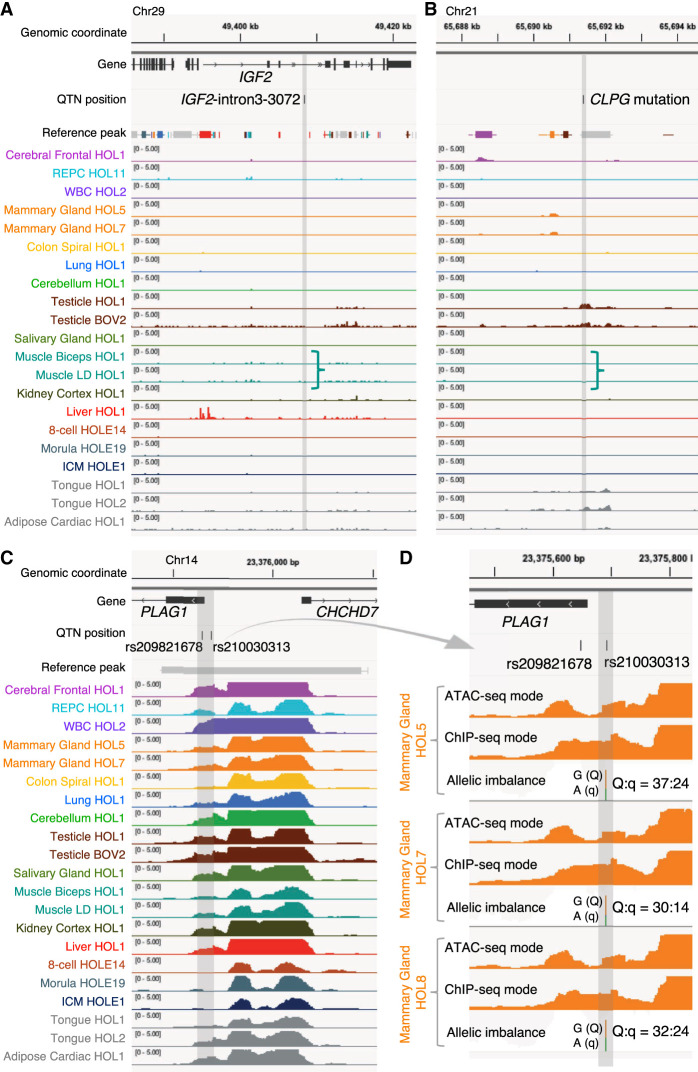
We report the generation of an organism-wide catalog of 976,813 cis-acting regulatory elements for the bovine detected by the assay for transposase accessible chromatin using sequencing (ATAC-seq). We regroup these regulatory elements in 16 components by nonnegative matrix factorization. Correlation between the genome-wide density of peaks and transcription start sites, correlation between peak accessibility and expression of neighboring genes, and enrichment in transcription factor binding motifs support their regulatory potential. Using a previously established catalog of 12,736,643 variants, we show that the proportion of single-nucleotide polymorphisms mapping to ATAC-seq peaks is higher than expected and that this is owing to an approximately 1.3-fold higher mutation rate within peaks. Their site frequency spectrum indicates that variants in ATAC-seq peaks are subject to purifying selection. We generate eQTL data sets for liver and blood and show that variants that drive eQTL fall into liver- and blood-specific ATAC-seq peaks more often than expected by chance. We combine ATAC-seq and eQTL data to estimate that the proportion of regulatory variants mapping to ATAC-seq peaks is approximately one in three and that the proportion of variants mapping to ATAC-seq peaks that are regulatory is approximately one in 25. We discuss the implication of these findings on the utility of ATAC-seq information to improve the accuracy of genomic selection.
© 2023 Yuan et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources