

Unraveling mucolipidosis type III gamma through whole genome sequencing in late-onset retinitis pigmentosa: a case report
- PMID: 37752499
- PMCID: PMC10523780
- DOI: 10.1186/s12886-023-03136-4
Unraveling mucolipidosis type III gamma through whole genome sequencing in late-onset retinitis pigmentosa: a case report
Abstract
Background: We describe the case of a 47-year-old man referred to a retinal clinic and diagnosed with late-onset retinitis pigmentosa. Surprisingly, genetic testing revealed compound heterozygous pathogenic variants in GNPTG, leading to the diagnosis of the autosomal recessive lysosomal storage disorder mucolipidosis type III gamma. Mucolipidosis type III gamma is typically diagnosed during childhood due to symptoms relating to skeletal dysplasia. Retinal dystrophy is not a common phenotypic feature.
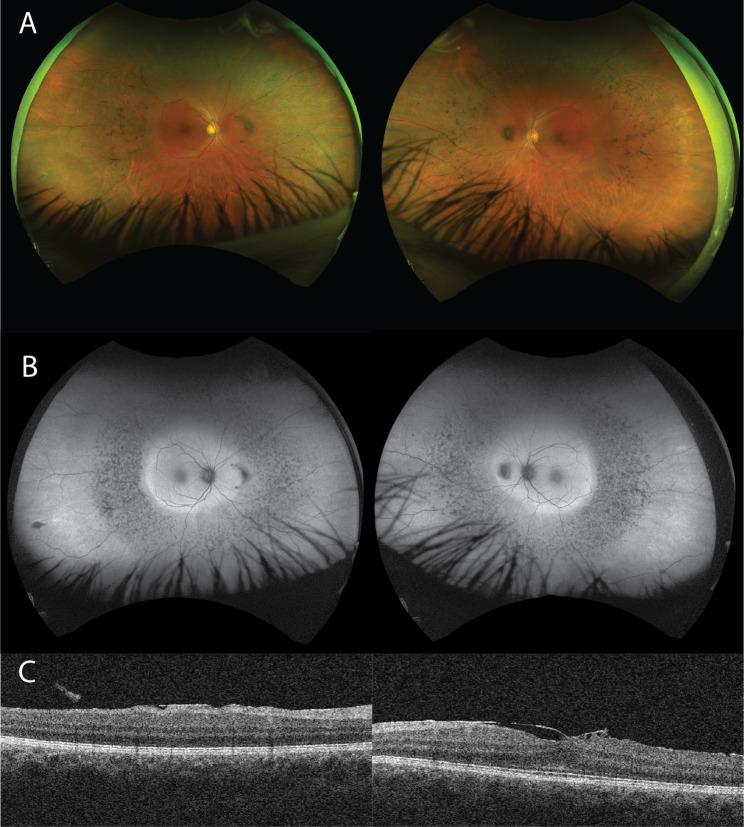
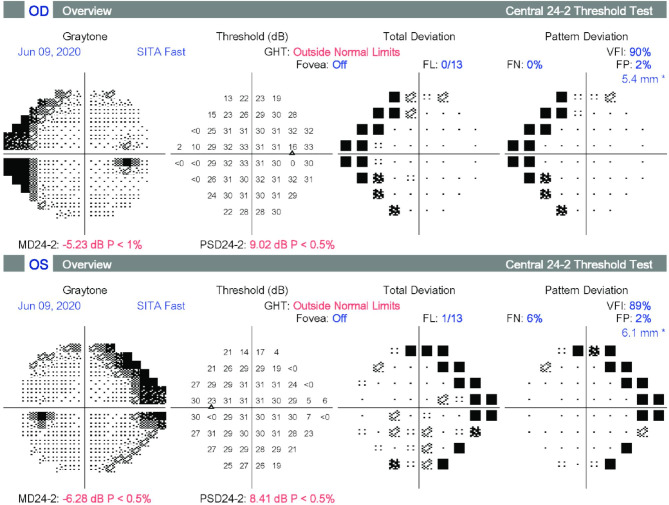
Case presentation: Ophthalmologic examination was consistent with a mild form of retinitis pigmentosa and included fundus photography, measurement of best-corrected visual acuity, optical coherence tomography, electroretinogram and visual field testing. Extraocular findings included joint restriction and pains from an early age leading to bilateral hip replacement by age 30, aortic insufficiency, and hypertension. Genetic analysis was performed by whole genome sequencing filtered for a gene panel of 325 genes associated with retinal disease. Two compound heterozygous pathogenic variants were identified in GNPTG, c.347_349del and c.607dup. The diagnosis of mucolipidosis type III gamma was confirmed biochemically by measurement of increased activities of specific lysosomal enzymes in plasma.
Conclusion: To our knowledge this is the first description of retinitis pigmentosa caused by compound heterozygous variants in GNPTG, providing further indications that late-onset retinal dystrophy is part of the phenotypic spectrum of mucolipidosis type III gamma.
Keywords: Case report; GNPTG; Inherited retinal dystrophy; Lysosomal disease; Mucolipidosis type III gamma; Retinitis pigmentosa; Whole genome sequencing.
© 2023. The Author(s).
Conflict of interest statement
KDG: None. KM: None. KN: None. ES: Receiver of research grant from Alnylam Pharmaceuticals Inc. AL: Member of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN-ITHACA [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516]. EB: Receiver of research grant from Novartis Sverige AB.
Figures
References
-
- Di Lorenzo G, Velho RV, Winter D, Thelen M, Ahmadi S, Schweizer M, et al. Lysosomal proteome and Secretome Analysis identifies Missorted enzymes and their nondegraded substrates in mucolipidosis III mouse cells. Mol Cell Proteomics. 2018;17(8):1612–26. doi: 10.1074/mcp.RA118.000720. - DOI - PMC - PubMed
-
- Varki AP, Reitman ML, Kornfeld S. Identification of a variant of mucolipidosis III (pseudo-hurler polydystrophy): a catalytically active N-acetylglucosaminylphosphotransferase that fails to phosphorylate lysosomal enzymes. Proc Natl Acad Sci USA. 1981;78(12):7773–7. doi: 10.1073/pnas.78.12.7773. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
