

Externalized histones fuel pulmonary fibrosis via a platelet-macrophage circuit of TGFβ1 and IL-27
- PMID: 37756334
- PMCID: PMC10556605
- DOI: 10.1073/pnas.2215421120
Externalized histones fuel pulmonary fibrosis via a platelet-macrophage circuit of TGFβ1 and IL-27
Abstract
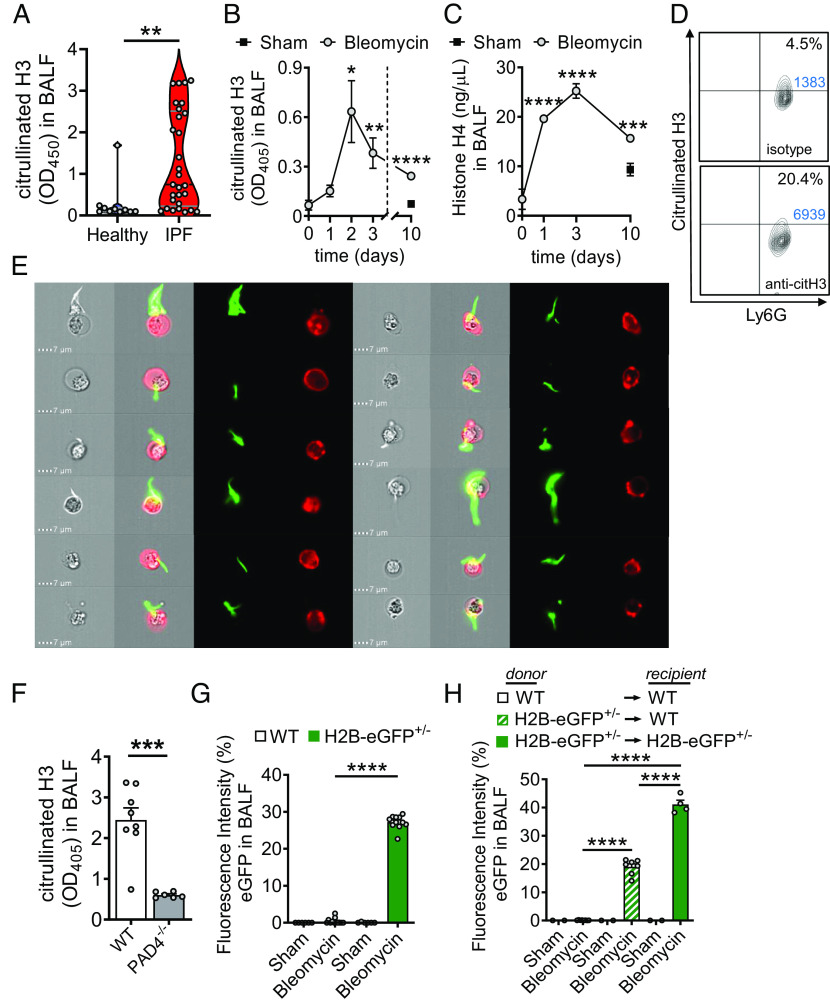
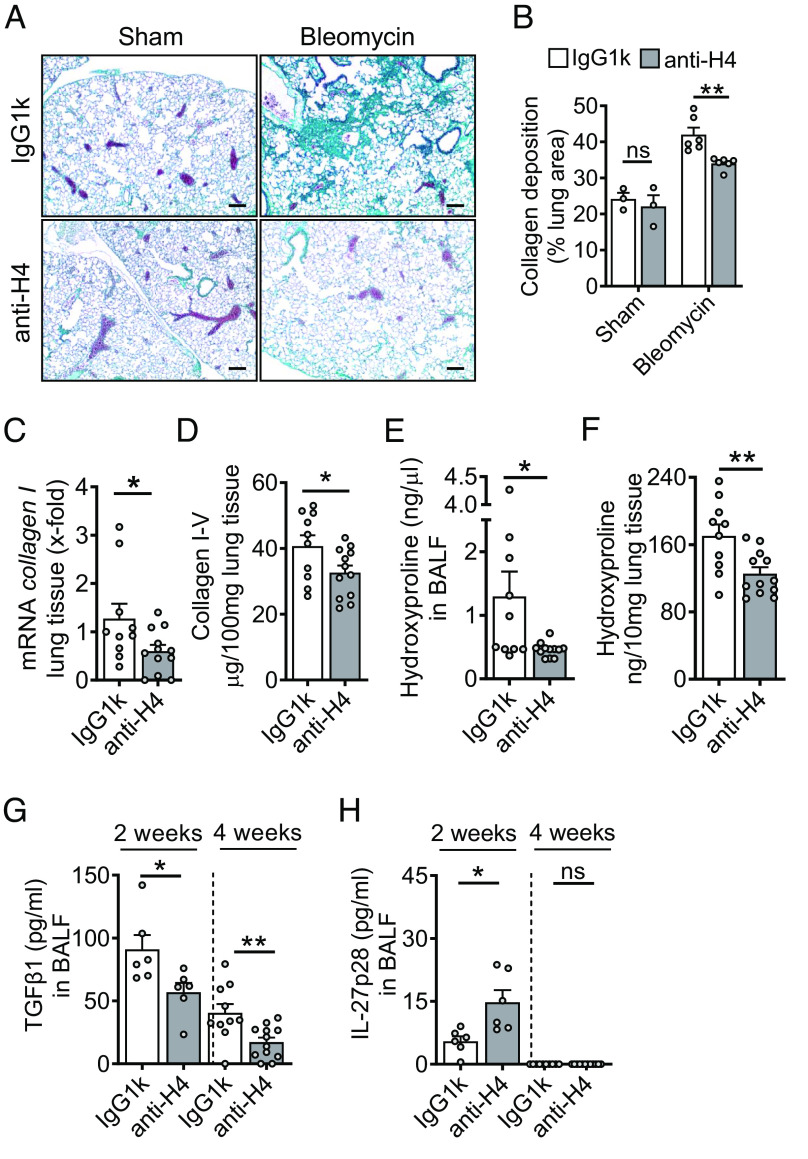
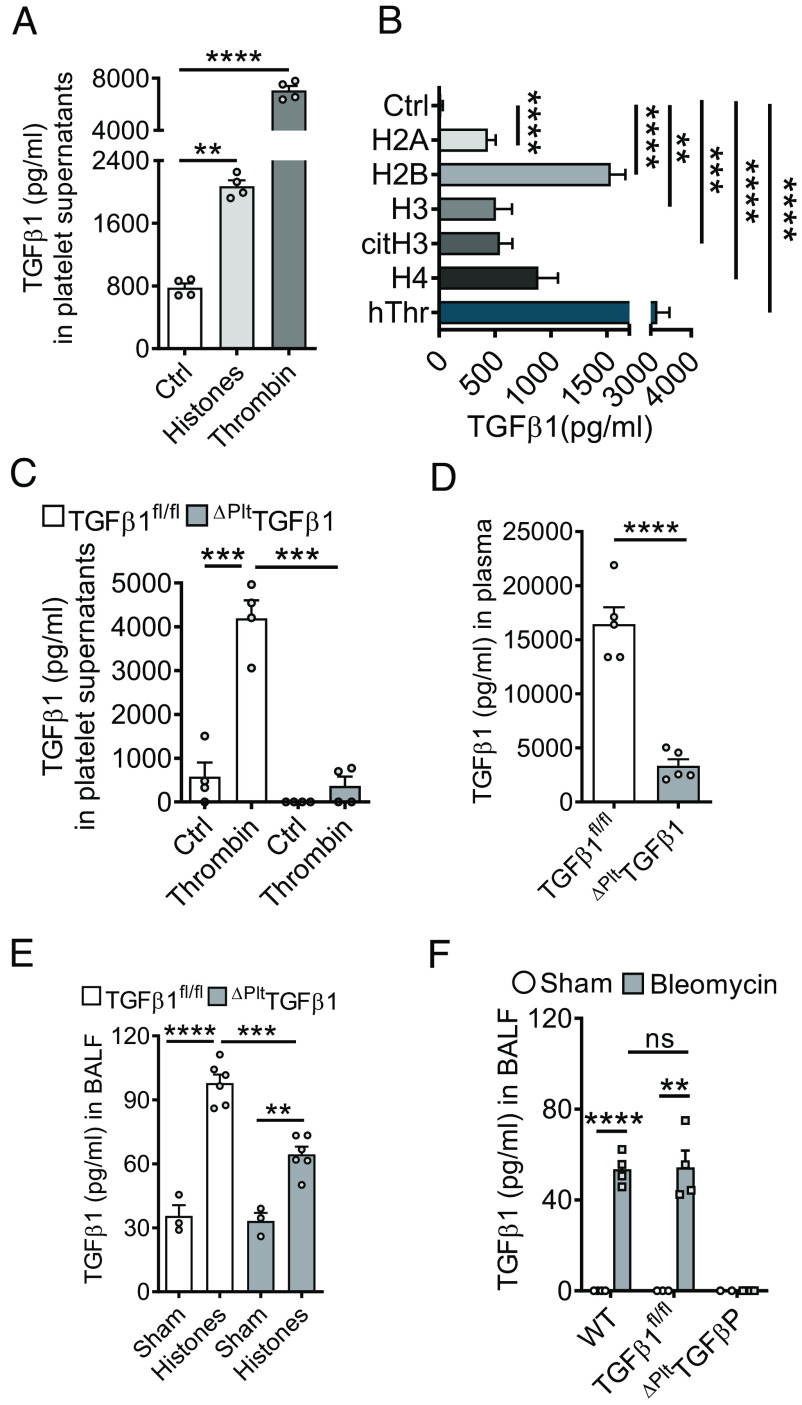
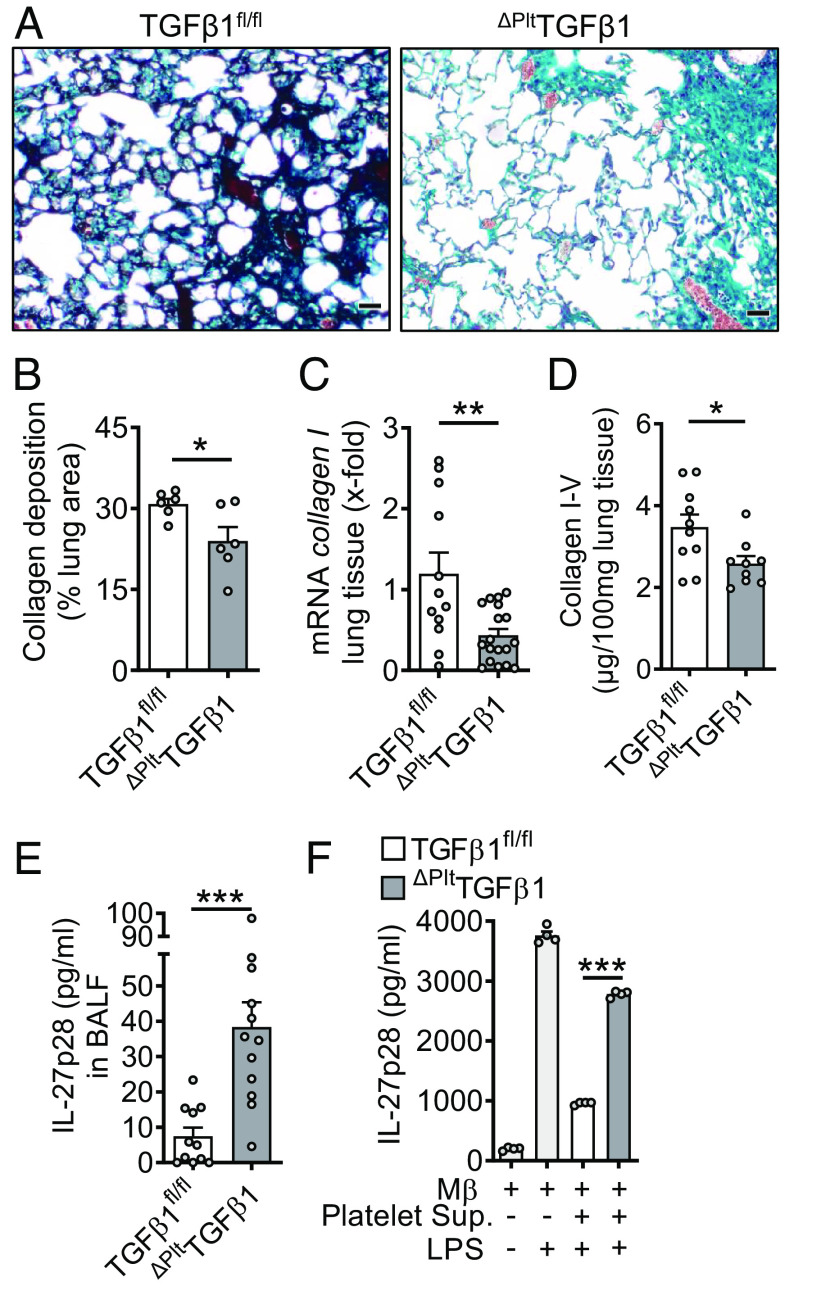
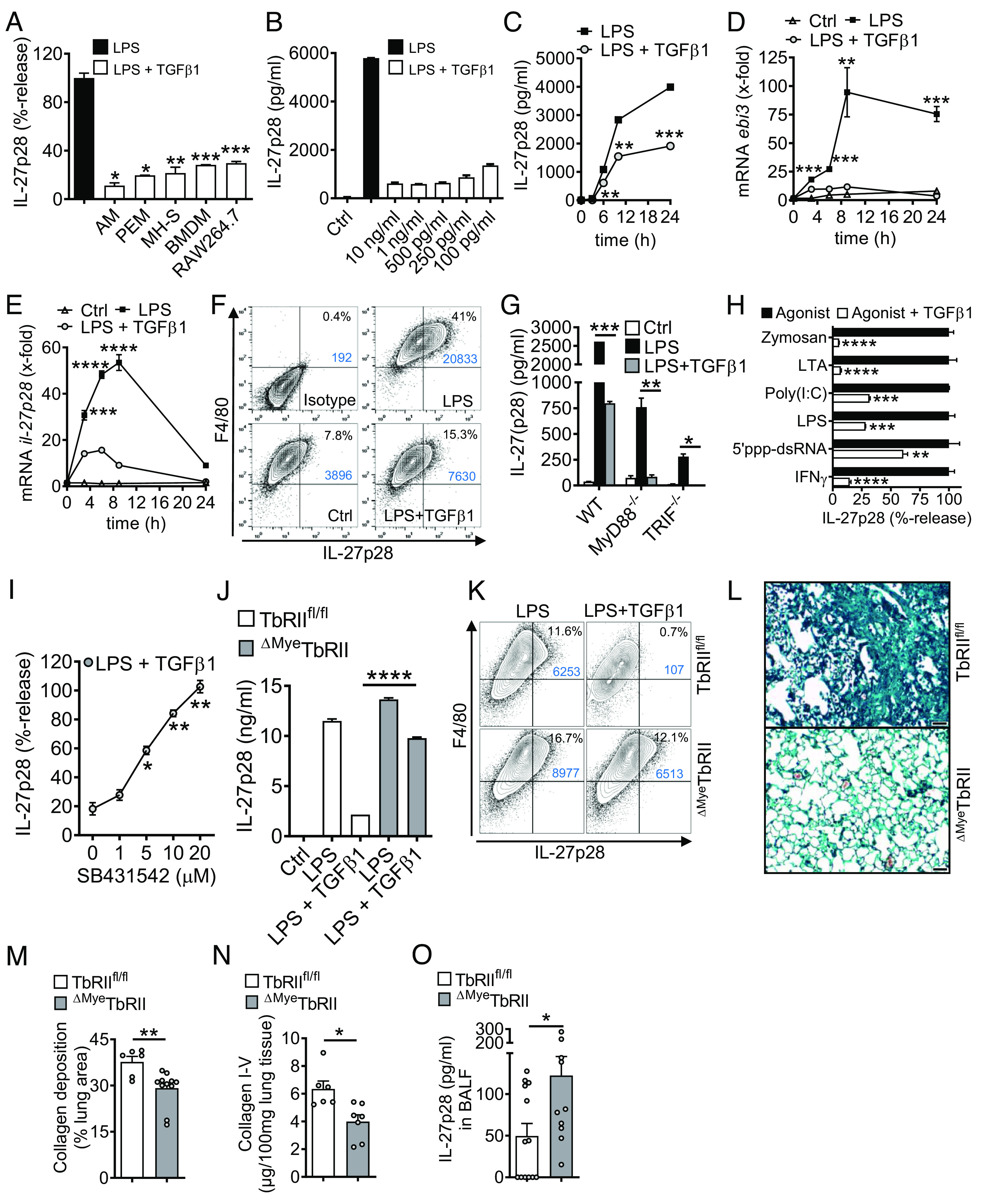
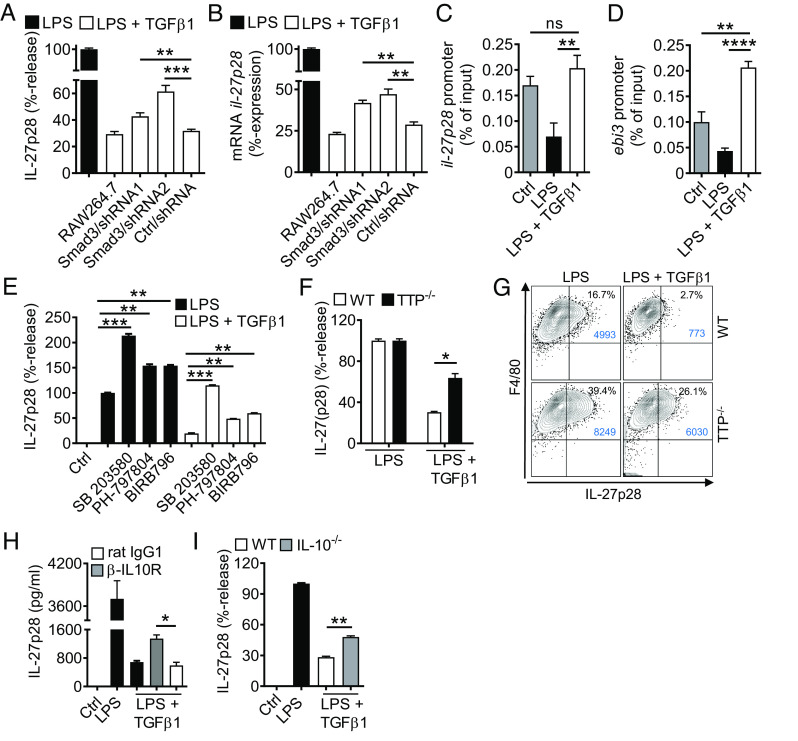
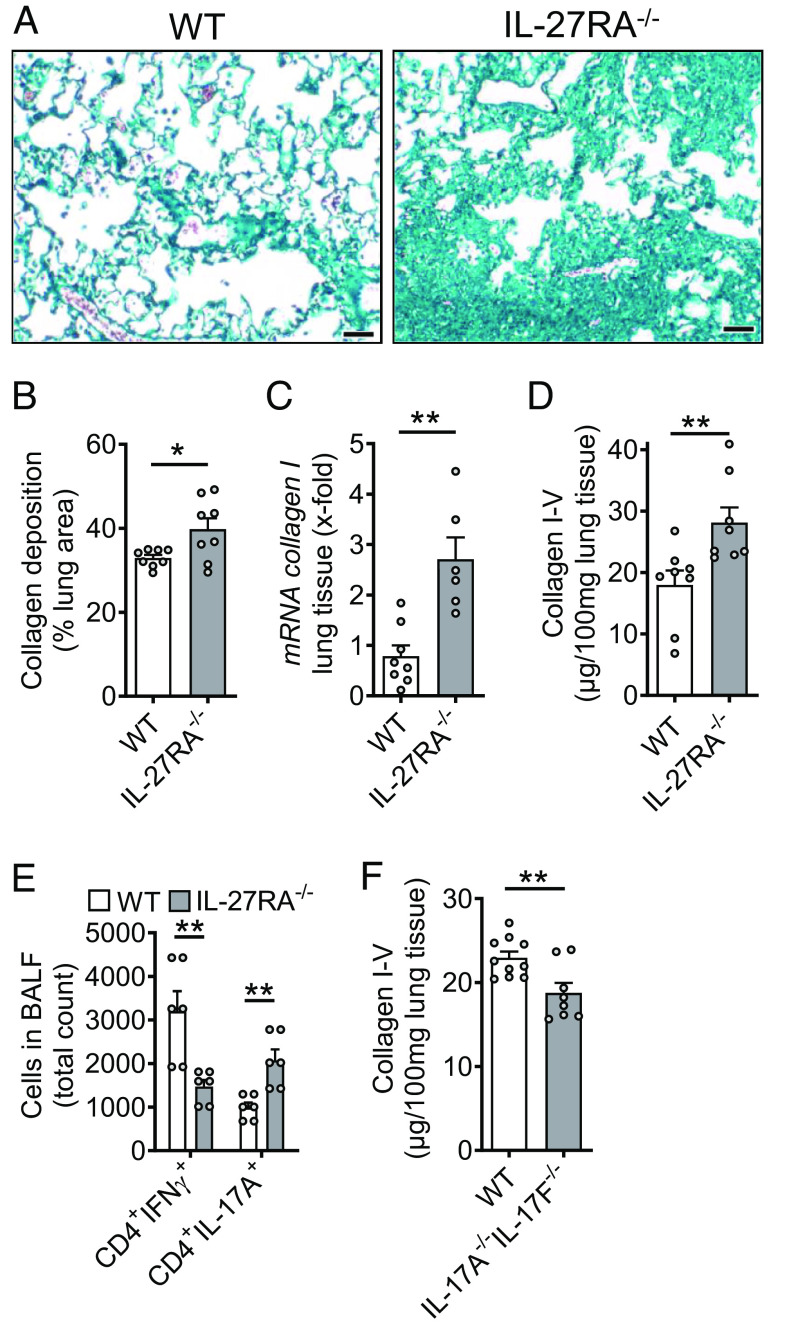
Externalized histones erupt from the nucleus as extracellular traps, are associated with several acute and chronic lung disorders, but their implications in the molecular pathogenesis of interstitial lung disease are incompletely defined. To investigate the role and molecular mechanisms of externalized histones within the immunologic networks of pulmonary fibrosis, we studied externalized histones in human and animal bronchoalveolar lavage (BAL) samples of lung fibrosis. Neutralizing anti-histone antibodies were administered in bleomycin-induced fibrosis of C57BL/6 J mice, and subsequent studies used conditional/constitutive knockout mouse strains for TGFβ and IL-27 signaling along with isolated platelets and cultured macrophages. We found that externalized histones (citH3) were significantly (P < 0.01) increased in cell-free BAL fluids of patients with idiopathic pulmonary fibrosis (IPF; n = 29) as compared to healthy controls (n = 10). The pulmonary sources of externalized histones were Ly6G+CD11b+ neutrophils and nonhematopoietic cells after bleomycin in mice. Neutralizing monoclonal anti-histone H2A/H4 antibodies reduced the pulmonary collagen accumulation and hydroxyproline concentration. Histones activated platelets to release TGFβ1, which signaled through the TGFbRI/TGFbRII receptor complex on LysM+ cells to antagonize macrophage-derived IL-27 production. TGFβ1 evoked multiple downstream mechanisms in macrophages, including p38 MAPK, tristetraprolin, IL-10, and binding of SMAD3 to the IL-27 promotor regions. IL-27RA-deficient mice displayed more severe collagen depositions suggesting that intact IL-27 signaling limits fibrosis. In conclusion, externalized histones inactivate a safety switch of antifibrotic, macrophage-derived IL-27 by boosting platelet-derived TGFβ1. Externalized histones are accessible to neutralizing antibodies for improving the severity of experimental pulmonary fibrosis.
Keywords: cytokines; innate immunity; macrophages; platelets; pulmonology.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Sgalla G., Kulkarni T., Antin-Ozerkis D., Thannickal V. J., Richeldi L., Update in pulmonary fibrosis 2018. Am. J. Respir. Crit. Care Med. 200, 292–300 (2019). - PubMed
-
- Smigiel K. S., Parks W. C., Macrophages, wound healing, and fibrosis: Recent insights. Curr. Rheumatol. Rep. 20, 17 (2018). - PubMed
-
- Leask A., Abraham D. J., TGF-beta signaling and the fibrotic response. FASEB J. 18, 816–827 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
