

Fundamental Neurochemistry Review: Microglial immunometabolism in traumatic brain injury
- PMID: 37759406
- PMCID: PMC10655864
- DOI: 10.1111/jnc.15959
Fundamental Neurochemistry Review: Microglial immunometabolism in traumatic brain injury
Abstract
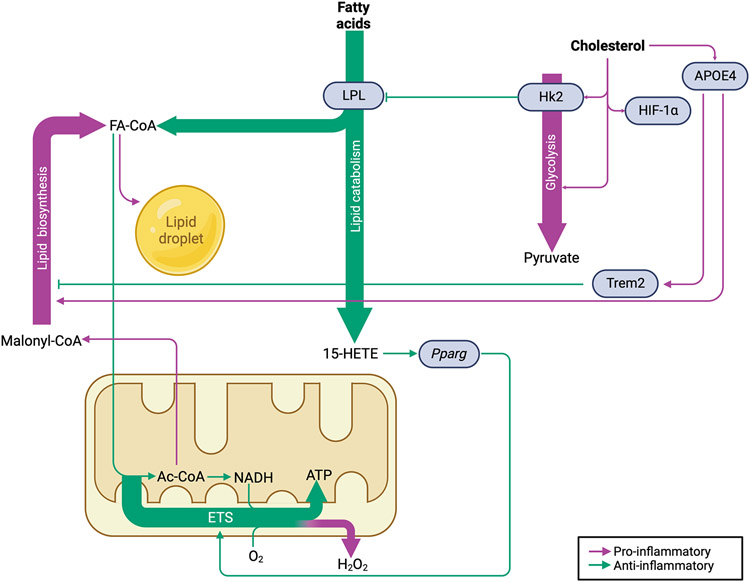
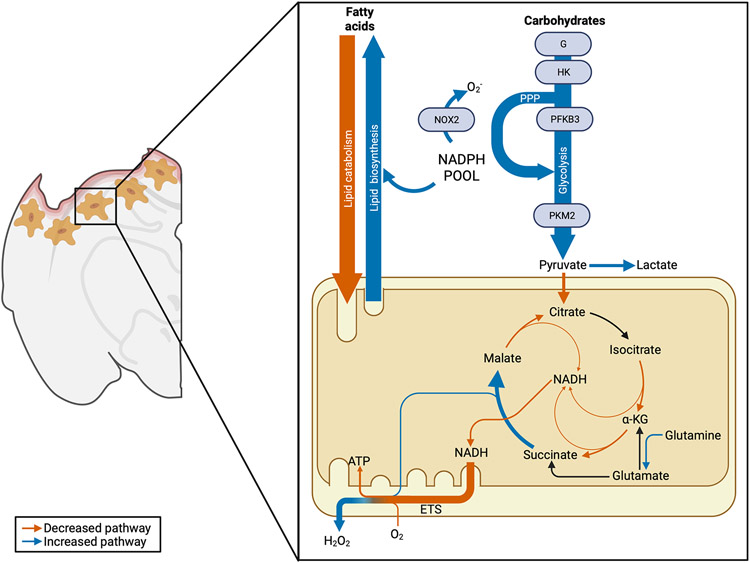
Traumatic brain injury (TBI) is a devastating neurological disorder caused by a physical impact to the brain that promotes diffuse damage and chronic neurodegeneration. Key mechanisms believed to support secondary brain injury include mitochondrial dysfunction and chronic neuroinflammation. Microglia and brain-infiltrating macrophages are responsible for neuroinflammatory cytokine and reactive oxygen species (ROS) production after TBI. Their production is associated with loss of homeostatic microglial functions such as immunosurveillance, phagocytosis, and immune resolution. Beyond providing energy support, mitochondrial metabolic pathways reprogram the pro- and anti-inflammatory machinery in immune cells, providing a critical immunometabolic axis capable of regulating immunologic response to noxious stimuli. In the brain, the capacity to adapt to different environmental stimuli derives, in part, from microglia's ability to recognize and respond to changes in extracellular and intracellular metabolite levels. This capacity is met by an equally plastic metabolism, capable of altering immune function. Microglial pro-inflammatory activation is associated with decreased mitochondrial respiration, whereas anti-inflammatory microglial polarization is supported by increased oxidative metabolism. These metabolic adaptations contribute to neuroimmune responses, placing mitochondria as a central regulator of post-traumatic neuroinflammation. Although it is established that profound neurometabolic changes occur following TBI, key questions related to metabolic shifts in microglia remain unresolved. These include (a) the nature of microglial mitochondrial dysfunction after TBI, (b) the hierarchical positions of different metabolic pathways such as glycolysis, pentose phosphate pathway, glutaminolysis, and lipid oxidation during secondary injury and recovery, and (c) how immunometabolism alters microglial phenotypes, culminating in chronic non-resolving neuroinflammation. In this basic neurochemistry review article, we describe the contributions of immunometabolism to TBI, detail primary evidence of mitochondrial dysfunction and metabolic impairments in microglia and macrophages, discuss how major metabolic pathways contribute to post-traumatic neuroinflammation, and set out future directions toward advancing immunometabolic phenotyping in TBI.
Keywords: metabolism; microglia; mitochondria; neuroimmunology; traumatic brain injury.
© 2023 The Authors. Journal of Neurochemistry published by John Wiley & Sons Ltd on behalf of International Society for Neurochemistry.
Conflict of interest statement
Conflict of interest:
The authors have no conflict of interest to declare.
Figures
Similar articles
-
Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits.J Neurosci. 2020 Apr 1;40(14):2960-2974. doi: 10.1523/JNEUROSCI.2402-19.2020. Epub 2020 Feb 24. J Neurosci. 2020. PMID: 32094203 Free PMC article.
-
Deletion of PTEN in microglia ameliorates chronic neuroinflammation following repetitive mTBI.Mol Cell Neurosci. 2023 Jun;125:103855. doi: 10.1016/j.mcn.2023.103855. Epub 2023 Apr 20. Mol Cell Neurosci. 2023. PMID: 37084991
-
Itaconate restrains acute proinflammatory activation of microglia after traumatic brain injury in mice.Sci Transl Med. 2025 Mar 12;17(789):eadn2635. doi: 10.1126/scitranslmed.adn2635. Epub 2025 Mar 12. Sci Transl Med. 2025. PMID: 40073156
-
Microglial NOX2 as a therapeutic target in traumatic brain injury: Mechanisms, consequences, and potential for neuroprotection.Ageing Res Rev. 2025 Jun;108:102735. doi: 10.1016/j.arr.2025.102735. Epub 2025 Mar 22. Ageing Res Rev. 2025. PMID: 40122395 Review.
-
Understanding microglial responses in large animal models of traumatic brain injury: an underutilized resource for preclinical and translational research.J Neuroinflammation. 2023 Mar 9;20(1):67. doi: 10.1186/s12974-023-02730-z. J Neuroinflammation. 2023. PMID: 36894951 Free PMC article. Review.
Cited by
-
Soluble Urokinase-Type Plasminogen Activator Receptor and Inflammatory Biomarker Response with Prognostic Significance after Acute Neuronal Injury - a Prospective Cohort Study.Inflammation. 2025 Aug;48(4):2217-2229. doi: 10.1007/s10753-024-02185-1. Epub 2024 Nov 14. Inflammation. 2025. PMID: 39540961 Free PMC article.
-
Mitophagy in acute central nervous system injuries: regulatory mechanisms and therapeutic potentials.Neural Regen Res. 2025 Sep 1;20(9):2437-2453. doi: 10.4103/NRR.NRR-D-24-00432. Epub 2024 Sep 6. Neural Regen Res. 2025. PMID: 39248161 Free PMC article.
-
High-dimensional proteomic analysis for pathophysiological classification of traumatic brain injury.Brain. 2025 Mar 6;148(3):1015-1030. doi: 10.1093/brain/awae305. Brain. 2025. PMID: 39323289 Free PMC article.
-
MRI-T2 Relaxometry is Increased in Mild Traumatic Brain Injury: Indications of Acute Brain Abnormalities After Injury.J Neurosci Res. 2025 Apr;103(4):e70034. doi: 10.1002/jnr.70034. J Neurosci Res. 2025. PMID: 40178334 Free PMC article.
-
Ischemic Stroke Induces ROS Accumulation, Maladaptive Mitophagy, and Neuronal Apoptosis in Minipigs.J Microbiol Biotechnol. 2024 Dec 28;34(12):2648-2661. doi: 10.4014/jmb.2409.09003. Epub 2024 Nov 14. J Microbiol Biotechnol. 2024. PMID: 39631782 Free PMC article.
References
-
- Almeida-Suhett C, Namboodiri AM, Clarke K and Deuster PA (2022) The ketone ester, 3-hydroxybutyl-3-hydroxybutyrate, attenuates neurobehavioral deficits and improves neuropathology following controlled cortical impact in male rats. Nutr Neurosci 25, 1287–1299. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
