

Let-7g Upregulation Attenuated the KRAS-PI3K-Rac1-Akt Axis-Mediated Bioenergetic Functions
- PMID: 37759534
- PMCID: PMC10527334
- DOI: 10.3390/cells12182313
Let-7g Upregulation Attenuated the KRAS-PI3K-Rac1-Akt Axis-Mediated Bioenergetic Functions
Abstract
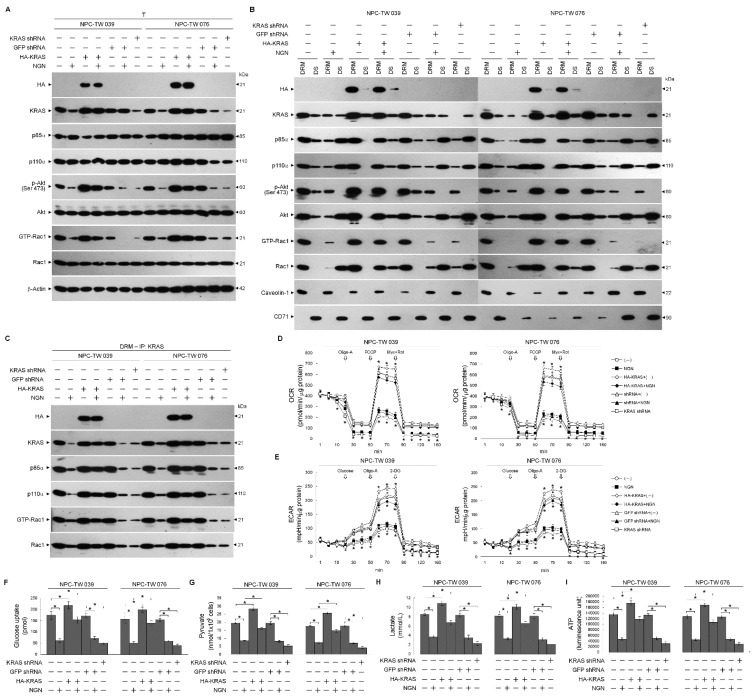
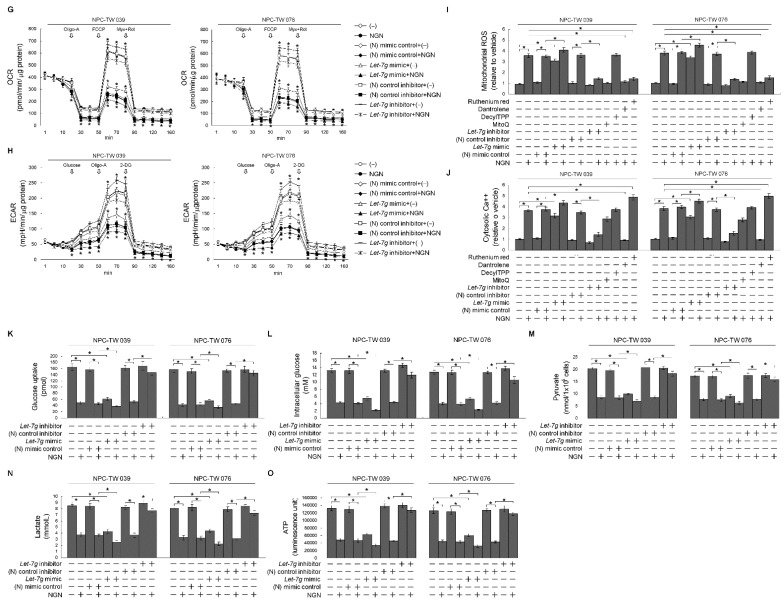
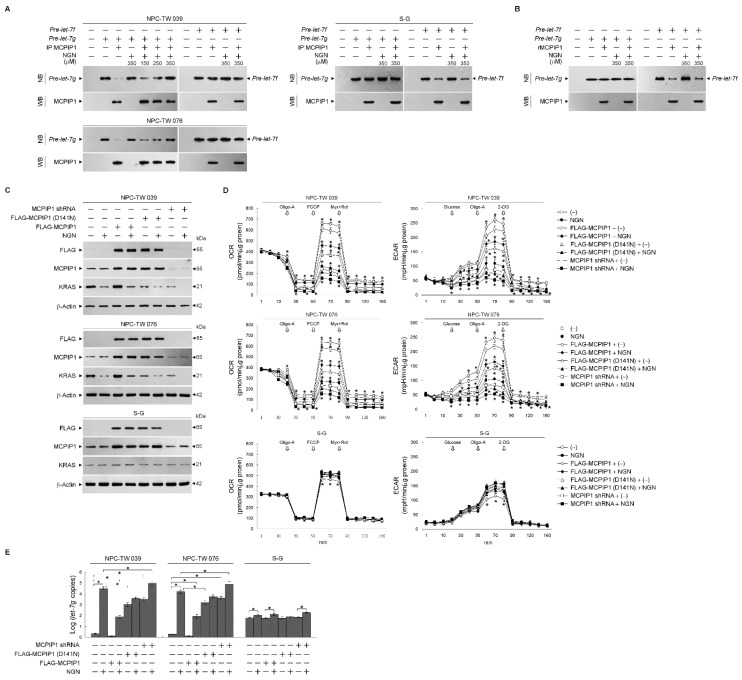
The aberrant activation of signaling pathways contributes to cancer cells with metabolic reprogramming. Thus, targeting signaling modulators is considered a potential therapeutic strategy for cancer. Subcellular fractionation, coimmunoprecipitation, biochemical analysis, and gene manipulation experiments revealed that decreasing the interaction of kirsten rat sarcoma viral oncogene homolog (KRAS) with p110α in lipid rafts with the use of naringenin (NGN), a citrus flavonoid, causes lipid raft-associated phosphatidylinositol 3-kinase (PI3K)-GTP-ras-related C3 botulinum toxin substrate 1 (Rac1)-protein kinase B (Akt)-regulated metabolic dysfunction of glycolysis and mitochondrial oxidative phosphorylation (OXPHOS), leading to apoptosis in human nasopharyngeal carcinoma (NPC) cells. The use of lethal-7g (let-7g) mimic and let-7g inhibitor confirmed that elevated let-7g resulted in a decrease in KRAS expression, which attenuated the PI3K-Rac1-Akt-BCL-2/BCL-xL-modulated mitochondrial energy metabolic functions. Increased let-7g depends on the suppression of the RNA-specificity of monocyte chemoattractant protein-induced protein-1 (MCPIP1) ribonuclease since NGN specifically blocks the degradation of pre-let-7g by NPC cell-derived immunoprecipitated MCPIP1. Converging lines of evidence indicate that the inhibition of MCPIP1 by NGN leads to let-7g upregulation, suppressing oncogenic KRAS-modulated PI3K-Rac1-Akt signaling and thereby impeding the metabolic activities of aerobic glycolysis and mitochondrial OXPHOS.
Keywords: KRAS; MCPIP1; bioenergetic metabolism; let-7g; naringenin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





Similar articles
-
Oxidative Stress-Induced Unscheduled CDK1-Cyclin B1 Activity Impairs ER-Mitochondria-Mediated Bioenergetic Metabolism.Cells. 2021 May 21;10(6):1280. doi: 10.3390/cells10061280. Cells. 2021. PMID: 34064109 Free PMC article.
-
Enforced C-Src Activation Causes Compartmental Dysregulation of PI3K and PTEN Molecules in Lipid Rafts of Tongue Squamous Carcinoma Cells by Attenuating Rac1-Akt-GLUT-1-Mediated Sphingolipid Synthesis.Int J Mol Sci. 2020 Aug 13;21(16):5812. doi: 10.3390/ijms21165812. Int J Mol Sci. 2020. PMID: 32823607 Free PMC article.
-
Suppressing the formation of lipid raft-associated Rac1/PI3K/Akt signaling complexes by curcumin inhibits SDF-1α-induced invasion of human esophageal carcinoma cells.Mol Carcinog. 2014 May;53(5):360-79. doi: 10.1002/mc.21984. Epub 2012 Nov 28. Mol Carcinog. 2014. PMID: 23192861
-
A single nuclear polymorphism in let-7g binding site affects the doubling time of thyroid nodule by regulating KRAS-induced cell proliferation.J Cell Physiol. 2019 Dec;234(12):23437-23447. doi: 10.1002/jcp.28912. Epub 2019 May 31. J Cell Physiol. 2019. PMID: 31152438
-
Targeting Mutant Kirsten Rat Sarcoma Viral Oncogene Homolog in Non-Small Cell Lung Cancer: Current Difficulties, Integrative Treatments and Future Perspectives.Front Pharmacol. 2022 Apr 20;13:875330. doi: 10.3389/fphar.2022.875330. eCollection 2022. Front Pharmacol. 2022. PMID: 35517800 Free PMC article. Review.
Cited by
-
Metabolic reprogramming in the pathogenesis and progression of nasopharyngeal carcinoma: molecular mechanisms and therapeutic implications.Am J Cancer Res. 2024 Aug 25;14(8):4049-4064. doi: 10.62347/VYAT9271. eCollection 2024. Am J Cancer Res. 2024. PMID: 39267663 Free PMC article. Review.
-
Contribution of Matrix Metalloproteinase-2 and Matrix Metalloproteinase-9 to Upper Tract Urothelial Cancer Risk in Taiwan.Life (Basel). 2024 Jun 26;14(7):801. doi: 10.3390/life14070801. Life (Basel). 2024. PMID: 39063556 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
