

Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View
- PMID: 37759540
- PMCID: PMC10527779
- DOI: 10.3390/cells12182318
Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View
Abstract
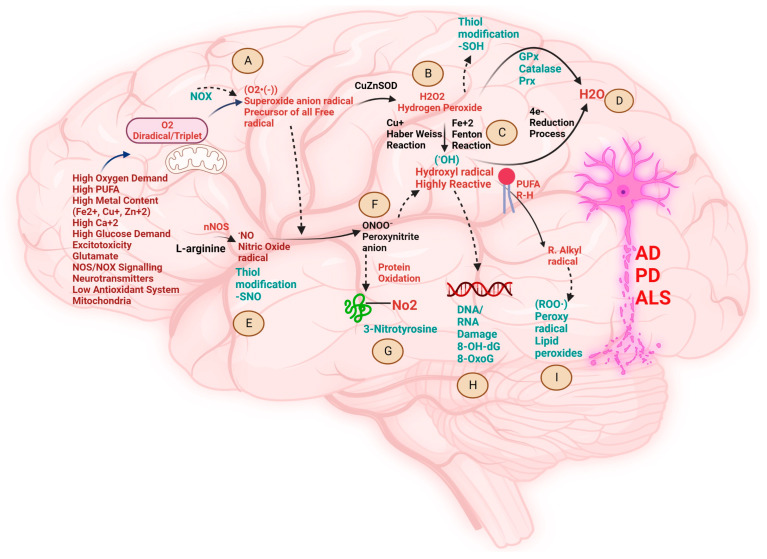
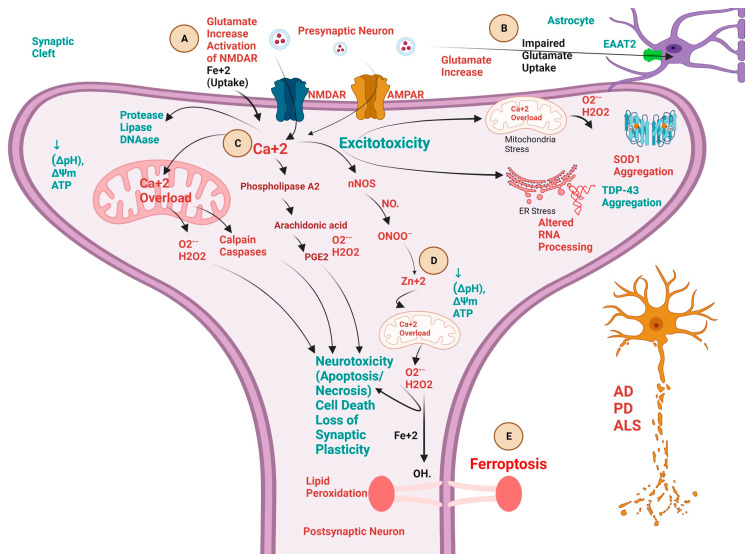
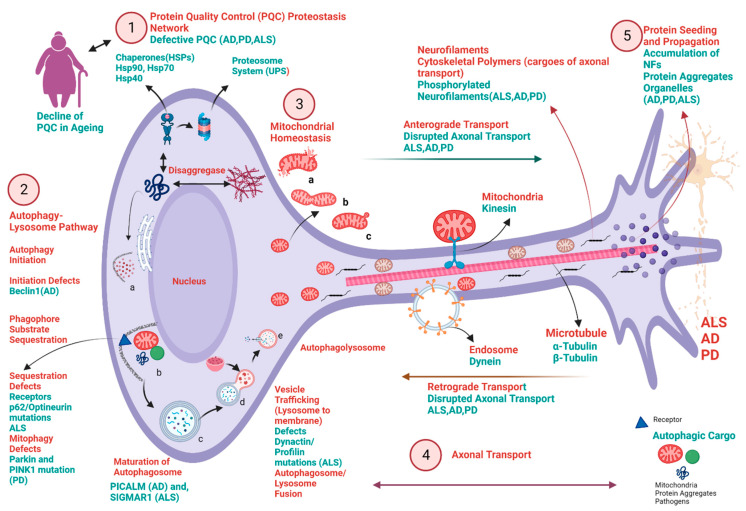
Neurodegenerative diseases (NDDs) like Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS) are defined by a myriad of complex aetiologies. Understanding the common biochemical molecular pathologies among NDDs gives an opportunity to decipher the overlapping and numerous cross-talk mechanisms of neurodegeneration. Numerous interrelated pathways lead to the progression of neurodegeneration. We present evidence from the past pieces of literature for the most usual global convergent hallmarks like ageing, oxidative stress, excitotoxicity-induced calcium butterfly effect, defective proteostasis including chaperones, autophagy, mitophagy, and proteosome networks, and neuroinflammation. Herein, we applied a holistic approach to identify and represent the shared mechanism across NDDs. Further, we believe that this approach could be helpful in identifying key modulators across NDDs, with a particular focus on AD, PD, and ALS. Moreover, these concepts could be applied to the development and diagnosis of novel strategies for diverse NDDs.
Keywords: Alzheimer’s disease; Parkinson’s disease; ageing; amyotrophic lateral sclerosis; autophagy; calcium butterfly effect; chaperones; excitotoxicity; mitophagy; neurodegenerative diseases; neuroinflammation; oxidative stress; proteostasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
