

Genomics of Wolfram Syndrome 1 (WFS1)
- PMID: 37759745
- PMCID: PMC10527379
- DOI: 10.3390/biom13091346
Genomics of Wolfram Syndrome 1 (WFS1)
Abstract
Wolfram Syndrome (WFS) is a rare, autosomal, recessive neurogenetic disorder that affects many organ systems. It is characterised by diabetes insipidus, diabetes mellites, optic atrophy, and deafness and, therefore, is also known as DIDMOAD. Nearly 15,000-30,000 people are affected by WFS worldwide, and, on average, patients suffering from WFS die at 30 years of age, usually from central respiratory failure caused by massive brain atrophy. The more prevalent of the two kinds of WFS is WFS1, which is a monogenic disease and caused by the loss of the WFS1 gene, whereas WFS2, which is more uncommon, is caused by mutations in the CISD2 gene. Currently, there is no treatment for WFS1 to increase the life expectancy of patients, and the treatments available do not significantly improve their quality of life. Understanding the genetics and the molecular mechanisms of WFS1 is essential to finding a cure. The inability of conventional medications to treat WFS1 points to the need for innovative strategies that must address the fundamental cause: the deletion of the WFS1 gene that leads to the profound ER stress and disturbances in proteostasis. An important approach here is to understand the mechanism of the cell degeneration after the deletion of the WFS1 gene and to describe the differences in these mechanisms for the different tissues. The studies so far have indicated that remarkable clinical heterogeneity is caused by the variable vulnerability caused by WFS1 mutations, and these differences cannot be attributed solely to the positions of mutations in the WFS1 gene. The present review gives a broader overview of the results from genomic studies on the WFS1 mouse model.
Keywords: WFS1 gene; Wolfram syndrome; functional genomics; genomics; wolframin protein.
Conflict of interest statement
The author declares no conflict of interest.
Figures
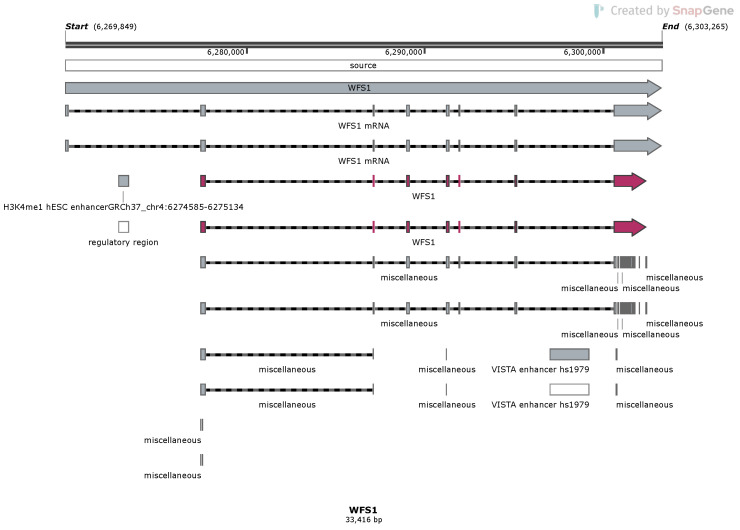
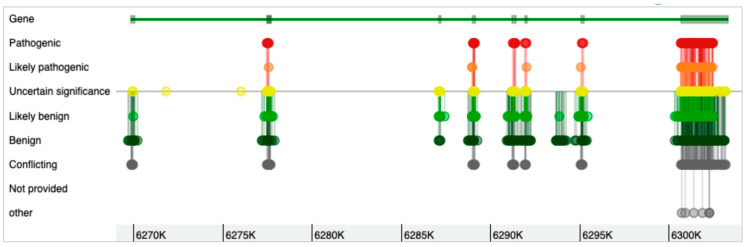
References
-
- Wolfram D.J., Wagener H.P. Diabetes Mellitus and Simple Optic Atrophy among Siblings: Report on Four Cases. Mayo Clin. Proc. 1938;13:715–718.
-
- Cremers C.W., Wijdeveld P.G., Pinckers A.J. Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). A review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr. Scand. Suppl. 1977;264:1–16. doi: 10.1111/j.1651-2227.1977.tb15069.x. - DOI - PubMed
-
- Inoue H., Tanizawa Y., Wasson J., Behn P., Kalidas K., Bernal-Mizrachi E., Mueckler M., Marshall H., Donis-Keller H., Crock P., et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) Nat. Genet. 1998;20:143–148. doi: 10.1038/2441. - DOI - PubMed
-
- Tanizawa Y., Inoue H., Oka Y. Positional cloning of the gene(WFS1) for Wolfram syndrome. Rinsho Byori. 2000;48:941–947. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
