

Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart
- PMID: 37759809
- PMCID: PMC10527470
- DOI: 10.3390/biom13091409
Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart
Abstract
Heart failure is a serious global health challenge, affecting more than 6.2 million people in the United States and is projected to reach over 8 million by 2030. Independent of etiology, failing hearts share common features, including defective calcium (Ca2+) handling, mitochondrial Ca2+ overload, and oxidative stress. In cardiomyocytes, Ca2+ not only regulates excitation-contraction coupling, but also mitochondrial metabolism and oxidative stress signaling, thereby controlling the function and actual destiny of the cell. Understanding the mechanisms of mitochondrial Ca2+ uptake and the molecular pathways involved in the regulation of increased mitochondrial Ca2+ influx is an ongoing challenge in order to identify novel therapeutic targets to alleviate the burden of heart failure. In this review, we discuss the mechanisms underlying altered mitochondrial Ca2+ handling in heart failure and the potential therapeutic strategies.
Keywords: calcium; heart failure; mitochondria.
Conflict of interest statement
Andrew R. Marks and Columbia University own shares in ARMGO Pharma, Inc., a biotechnology company developing RyR targeted drugs. All the remaining authors declare no conflict of interest.
Figures
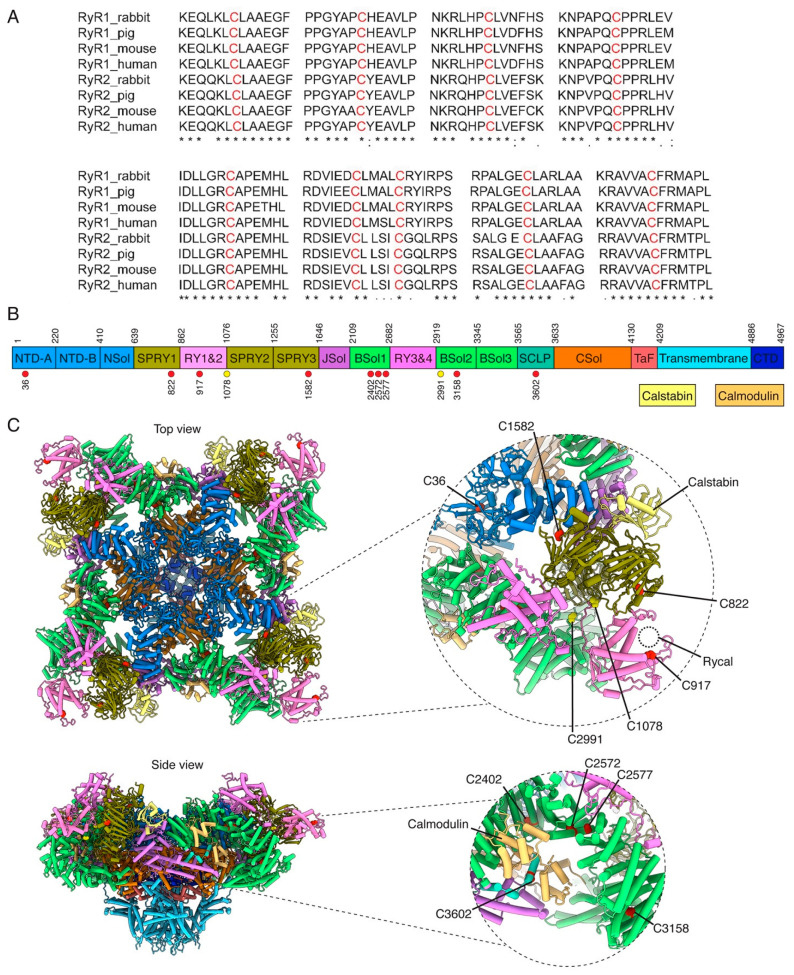
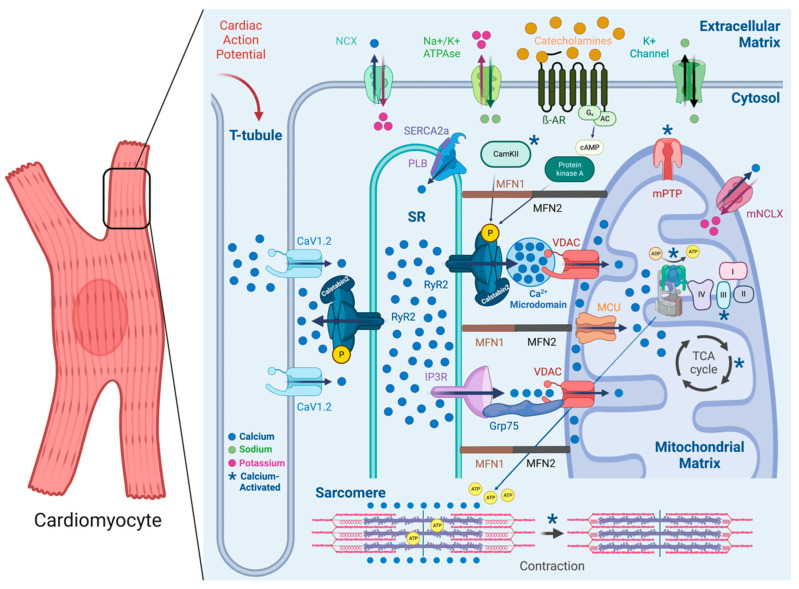
References
-
- Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. - DOI - PubMed
-
- Santulli G. Pathophysiology and Pharmacotherapy of Cardiovascular Disease. Adis; Cham, Switzerland: 2015. Sympathetic nervous system signaling in heart failure and cardiac aging; pp. 83–105. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
