

Thioredoxin/Glutaredoxin Systems and Gut Microbiota in NAFLD: Interplay, Mechanism, and Therapeutical Potential
- PMID: 37759983
- PMCID: PMC10525532
- DOI: 10.3390/antiox12091680
Thioredoxin/Glutaredoxin Systems and Gut Microbiota in NAFLD: Interplay, Mechanism, and Therapeutical Potential
Abstract
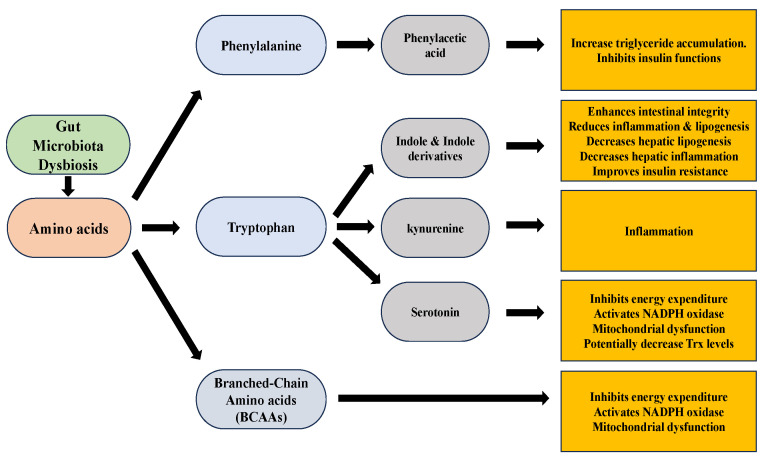
Non-alcoholic fatty liver disease (NAFLD) is a common clinical disease, and its pathogenesis is closely linked to oxidative stress and gut microbiota dysbiosis. Recently accumulating evidence indicates that the thioredoxin and glutaredoxin systems, the two thiol-redox dependent antioxidant systems, are the key players in the NAFLD's development and progression. However, the effects of gut microbiota dysbiosis on the liver thiol-redox systems are not well clarified. This review explores the role and mechanisms of oxidative stress induced by bacteria in NAFLD while emphasizing the crucial interplay between gut microbiota dysbiosis and Trx mediated-redox regulation. The paper explores how dysbiosis affects the production of specific gut microbiota metabolites, such as trimethylamine N-oxide (TMAO), lipopolysaccharides (LPS), short-chain fatty acids (SCFAs), amino acids, bile acid, and alcohol. These metabolites, in turn, significantly impact liver inflammation, lipid metabolism, insulin resistance, and cellular damage through thiol-dependent redox signaling. It suggests that comprehensive approaches targeting both gut microbiota dysbiosis and the thiol-redox antioxidant system are essential for effectively preventing and treating NAFLD. Overall, comprehending the intricate relationship between gut microbiota dysbiosis and thiol-redox systems in NAFLD holds significant promise in enhancing patient outcomes and fostering the development of innovative therapeutic interventions.
Keywords: NAFLD; gut microbiota dysbiosis; oxidative stress; reactive oxygen species; thioredoxin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
