

VprBP/DCAF1 Triggers Melanomagenic Gene Silencing through Histone H2A Phosphorylation
- PMID: 37760992
- PMCID: PMC10526264
- DOI: 10.3390/biomedicines11092552
VprBP/DCAF1 Triggers Melanomagenic Gene Silencing through Histone H2A Phosphorylation
Abstract
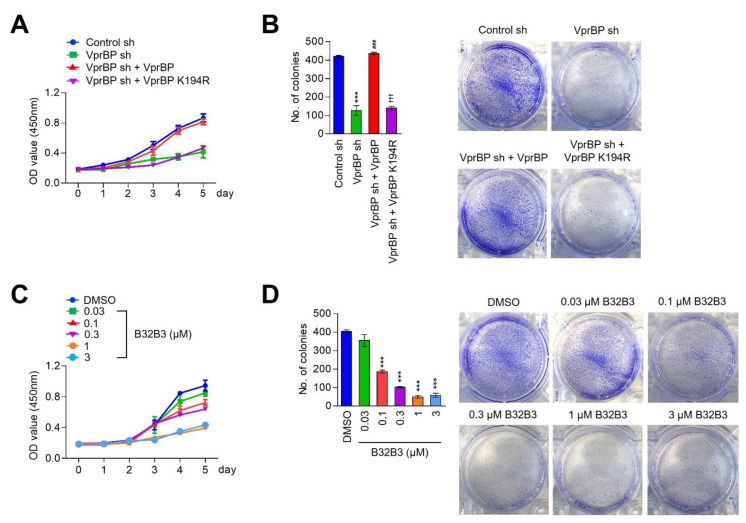
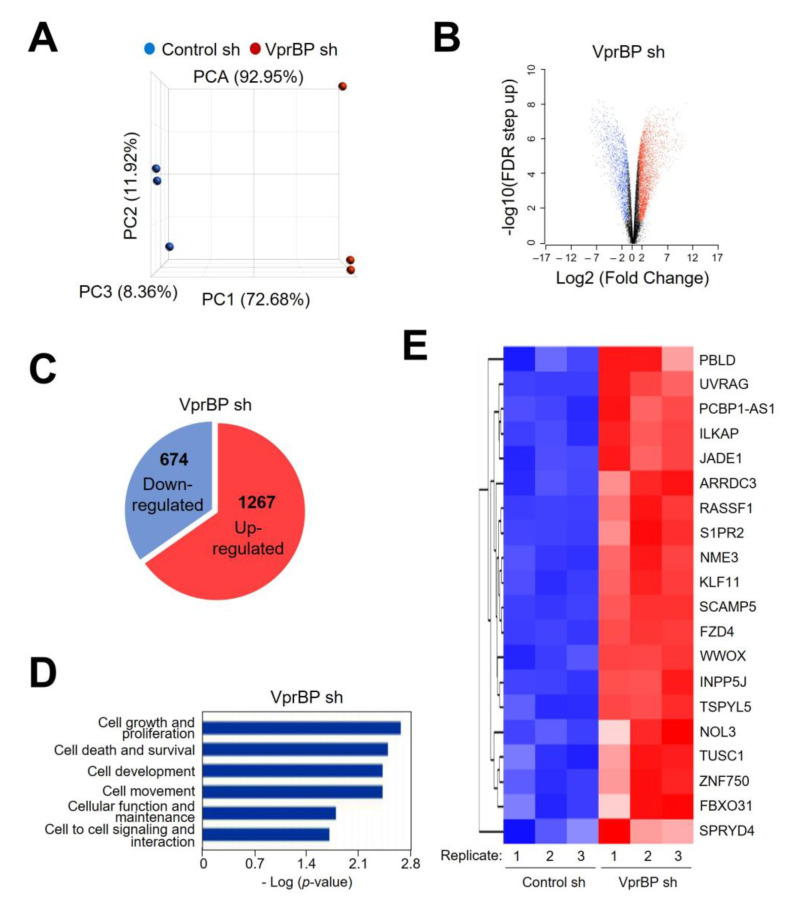
Vpr binding protein (VprBP), also known as DDB1- and CUL4-associated factor1 (DCAF1), is a recently identified atypical kinase and plays an important role in downregulating the transcription of tumor suppressor genes as well as increasing the risk for colon and prostate cancers. Melanoma is the most aggressive form of skin cancer arising from pigment-producing melanocytes and is often associated with the dysregulation of epigenetic factors targeting histones. Here, we demonstrate that VprBP is highly expressed and phosphorylates threonine 120 (T120) on histone H2A to drive the transcriptional inactivation of growth-regulatory genes in melanoma cells. As is the case for its epigenetic function in other types of cancers, VprBP acts to induce a gene silencing program dependent on H2AT120 phosphorylation (H2AT120p). The significance of VprBP-mediated H2AT120p is further underscored by the fact that VprBP knockdown- or VprBP inhibitor-induced lockage of H2AT120p mitigates melanoma tumor growth in xenograft models. Collectively, our results establish VprBP-mediated H2AT120p as a key epigenetic signal for melanomagenesis and suggest the therapeutic potential of targeting VprBP kinase activity for effective melanoma treatment.
Keywords: H2A; VprBP; histone; kinase; melanoma; phosphorylation.
Conflict of interest statement
The authors declare no competing interests.
Figures





Update of
-
VprBP/DCAF1 triggers melanomagenic gene silencing through histone H2A phosphorylation.Res Sq [Preprint]. 2023 Jul 12:rs.3.rs-2950076. doi: 10.21203/rs.3.rs-2950076/v2. Res Sq. 2023. Update in: Biomedicines. 2023 Sep 17;11(9):2552. doi: 10.3390/biomedicines11092552. PMID: 37293029 Free PMC article. Updated. Preprint.
Similar articles
-
VprBP/DCAF1 triggers melanomagenic gene silencing through histone H2A phosphorylation.Res Sq [Preprint]. 2023 Jul 12:rs.3.rs-2950076. doi: 10.21203/rs.3.rs-2950076/v2. Res Sq. 2023. Update in: Biomedicines. 2023 Sep 17;11(9):2552. doi: 10.3390/biomedicines11092552. PMID: 37293029 Free PMC article. Updated. Preprint.
-
VprBP directs epigenetic gene silencing through histone H2A phosphorylation in colon cancer.Mol Oncol. 2021 Oct;15(10):2801-2817. doi: 10.1002/1878-0261.13068. Epub 2021 Aug 8. Mol Oncol. 2021. PMID: 34312968 Free PMC article.
-
VprBP has intrinsic kinase activity targeting histone H2A and represses gene transcription.Mol Cell. 2013 Nov 7;52(3):459-67. doi: 10.1016/j.molcel.2013.09.017. Epub 2013 Oct 17. Mol Cell. 2013. PMID: 24140421 Free PMC article.
-
Histone H2A ubiquitination in transcriptional regulation and DNA damage repair.Int J Biochem Cell Biol. 2009 Jan;41(1):12-5. doi: 10.1016/j.biocel.2008.09.016. Epub 2008 Sep 26. Int J Biochem Cell Biol. 2009. PMID: 18929679 Review.
-
VprBP (DCAF1): a promiscuous substrate recognition subunit that incorporates into both RING-family CRL4 and HECT-family EDD/UBR5 E3 ubiquitin ligases.BMC Mol Biol. 2013 Sep 13;14:22. doi: 10.1186/1471-2199-14-22. BMC Mol Biol. 2013. PMID: 24028781 Free PMC article. Review.
Cited by
-
Functions and mechanisms of non-histone post-translational modifications in cancer progression.Cell Death Discov. 2025 Mar 31;11(1):125. doi: 10.1038/s41420-025-02410-2. Cell Death Discov. 2025. PMID: 40164592 Free PMC article. Review.
-
[Public Database-based Study to Explore the Expression and Role of DDB1 in Lung Adenocarcinoma].Zhongguo Fei Ai Za Zhi. 2025 Apr 20;28(4):256-266. doi: 10.3779/j.issn.1009-3419.2025.102.12. Zhongguo Fei Ai Za Zhi. 2025. PMID: 40404474 Free PMC article. Chinese.
-
MMP-9-dependent proteolysis of the histone H3 N-terminal tail: a critical epigenetic step in driving oncogenic transcription and colon tumorigenesis.Mol Oncol. 2024 Aug;18(8):2001-2019. doi: 10.1002/1878-0261.13652. Epub 2024 Apr 10. Mol Oncol. 2024. PMID: 38600695 Free PMC article.
-
Glucose deprivation triggers DCAF1-mediated inactivation of Rheb-mTORC1 and promotes cancer cell survival.Cell Death Dis. 2024 Jun 11;15(6):409. doi: 10.1038/s41419-024-06808-1. Cell Death Dis. 2024. PMID: 38862475 Free PMC article.
References
-
- McCall C.M., de Marval P.L.M., Chastain P.D., 2nd, Jackson S.C., He Y.J., Kotake Y., Cook J.G., Xiong Y. Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol. Cell. Biol. 2008;28:5621–5633. doi: 10.1128/MCB.00232-08. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
