

Snijders Blok-Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review
- PMID: 37761804
- PMCID: PMC10530855
- DOI: 10.3390/genes14091664
Snijders Blok-Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review
Abstract
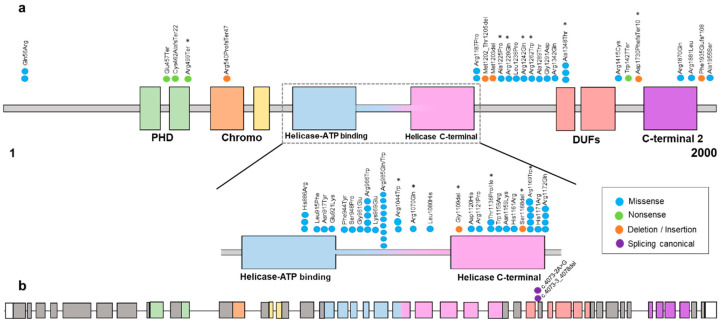
Snijders Blok-Campeau syndrome (SNIBCPS, OMIM# 618205) is an extremely infrequent disease with only approximately 60 cases reported so far. SNIBCPS belongs to the group of neurodevelopmental disorders (NDDs). Clinical features of patients with SNIBCPS include global developmental delay, intellectual disability, speech and language difficulties and behavioral disorders like autism spectrum disorder. In addition, patients with SNIBCPS exhibit typical dysmorphic features including macrocephaly, hypertelorism, sparse eyebrows, broad forehead, prominent nose and pointed chin. The severity of the neurological effects as well as the presence of other features is variable among subjects. SNIBCPS is caused likely by pathogenic and pathogenic variants in CHD3 (Chromodomain Helicase DNA Binding Protein 3), which seems to be involved in chromatin remodeling by deacetylating histones. Here, we report 20 additional patients with clinical features compatible with SNIBCPS from 17 unrelated families with confirmed likely pathogenic/pathogenic variants in CHD3. Patients were analyzed by whole exome sequencing and segregation studies were performed by Sanger sequencing. Patients in this study showed different pathogenic variants affecting several functional domains of the protein. Additionally, none of the variants described here were reported in control population databases, and most computational predictors suggest that they are deleterious. The most common clinical features of the whole cohort of patients are global developmental delay (98%) and speech disorder/delay (92%). Other frequent features (51-74%) include intellectual disability, hypotonia, hypertelorism, abnormality of vision, macrocephaly and prominent forehead, among others. This study expands the number of individuals with confirmed SNIBCPS due to pathogenic or likely pathogenic variants in CHD3. Furthermore, we add evidence of the importance of the application of massive parallel sequencing for NDD patients for whom the clinical diagnosis might be challenging and where deep phenotyping is extremely useful to accurately manage and follow up the patients.
Keywords: CHD3; SNIBCPS; Snijders Blok–Campeau syndrome; neurodevelopmental disorders; overgrowth.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Snijders Blok L., Rousseau J., Twist J., Ehresmann S., Takaku M., Venselaar H., Rodan L.H., Nowak C.B., Douglas J., Swoboda K.J., et al. CHD3 Helicase Domain Mutations Cause a Neurodevelopmental Syndrome with Macrocephaly and Impaired Speech and Language. Nat. Commun. 2018;9:4619. doi: 10.1038/s41467-018-06014-6. - DOI - PMC - PubMed
-
- Drivas T.G., Li D., Nair D., Alaimo J.T., Alders M., Altmüller J., Barakat T.S., Bebin E.M., Bertsch N.L., Blackburn P.R., et al. A Second Cohort of CHD3 Patients Expands the Molecular Mechanisms Known to Cause Snijders Blok-Campeau Syndrome. Eur. J. Hum. Genet. 2020;28:1422–1431. doi: 10.1038/s41431-020-0654-4. - DOI - PMC - PubMed
-
- Coursimault J., Lecoquierre F., Saugier-Veber P., Drouin-Garraud V., Lechevallier J., Boland A., Deleuze J.-F., Frebourg T., Nicolas G., Brehin A.-C. Hypersociability Associated with Developmental Delay, Macrocephaly and Facial Dysmorphism Points to CHD3 Mutations. Eur. J. Med. Genet. 2021;64:104166. doi: 10.1016/j.ejmg.2021.104166. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
