

Genetic Modifiers of Mendelian Monogenic Collagen IV Nephropathies in Humans and Mice
- PMID: 37761826
- PMCID: PMC10530214
- DOI: 10.3390/genes14091686
Genetic Modifiers of Mendelian Monogenic Collagen IV Nephropathies in Humans and Mice
Abstract
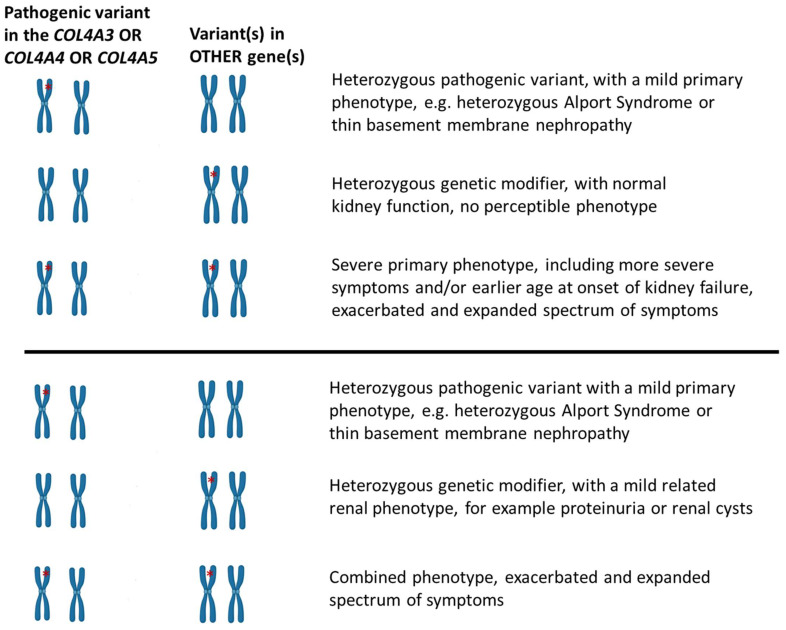
Familial hematuria is a clinical sign of a genetically heterogeneous group of conditions, accompanied by broad inter- and intrafamilial variable expressivity. The most frequent condition is caused by pathogenic (or likely pathogenic) variants in the collagen-IV genes, COL4A3/A4/A5. Pathogenic variants in COL4A5 are responsible for the severe X-linked glomerulopathy, Alport syndrome (AS), while homozygous or compound heterozygous variants in the COL4A3 or the COL4A4 gene cause autosomal recessive AS. AS usually leads to progressive kidney failure before the age of 40-years when left untreated. People who inherit heterozygous COL4A3/A4 variants are at-risk of a slowly progressive form of the disease, starting with microscopic hematuria in early childhood, developing Alport spectrum nephropathy. Sometimes, they are diagnosed with benign familial hematuria, and sometimes with autosomal dominant AS. At diagnosis, they often show thin basement membrane nephropathy, reflecting the uniform thin glomerular basement membrane lesion, inherited as an autosomal dominant condition. On a long follow-up, most patients will retain normal or mildly affected kidney function, while a substantial proportion will develop chronic kidney disease (CKD), even kidney failure at an average age of 55-years. A question that remains unanswered is how to distinguish those patients with AS or with heterozygous COL4A3/A4 variants who will manifest a more aggressive kidney function decline, requiring prompt medical intervention. The hypothesis that a subgroup of patients coinherit additional genetic modifiers that exacerbate their clinical course has been investigated by several researchers. Here, we review all publications that describe the potential role of candidate genetic modifiers in patients and include a summary of studies in AS mouse models.
Keywords: Alport syndrome; COL4 nephropathies; focal segmental glomerulosclerosis; genetic modifiers; glomerular diseases; thin basement membrane nephropathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Gross O., Kashtan C.E., Rheault M.N., Flinter F., Savige J., Miner J.H., Torra R., Ars E., Deltas C., Savva I., et al. Advances and unmet needs in genetic, basic and clinical science in Alport syndrome: Report from the 2015 International Workshop on Alport Syndrome. Nephrol. Dial. Transplant. 2016;32:916–924. doi: 10.1093/ndt/gfw095. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
