

Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors
- PMID: 37761873
- PMCID: PMC10530651
- DOI: 10.3390/genes14091733
Mechanisms of Regulation of the CHRDL1 Gene by the TWIST2 and ADD1/SREBP1c Transcription Factors
Abstract
Setleis syndrome (SS) is a rare focal facial dermal dysplasia caused by recessive mutations in the basic helix-loop-helix (bHLH) transcription factor, TWIST2. Expression microarray analysis showed that the chordin-like 1 (CHRDL1) gene is up-regulated in dermal fibroblasts from three SS patients with the Q119X TWIST2 mutation.
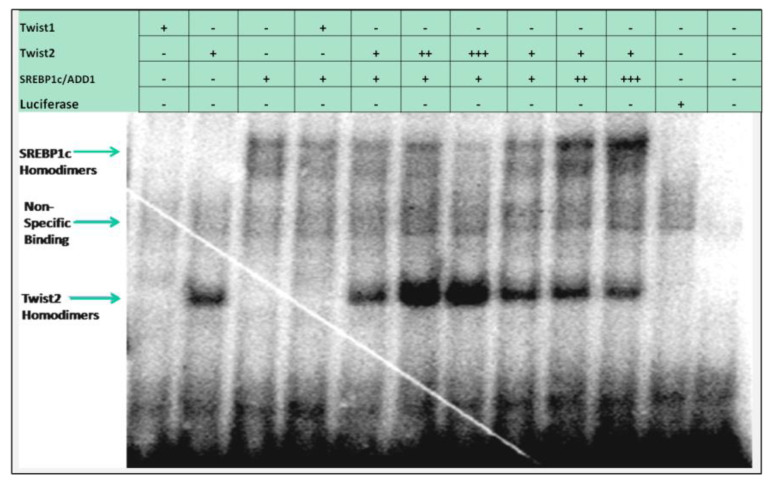
Methods: Putative TWIST binding sites were found in the upstream region of the CHRDL1 gene and examined by electrophoretic mobility shift (EMSA) and reporter gene assays.
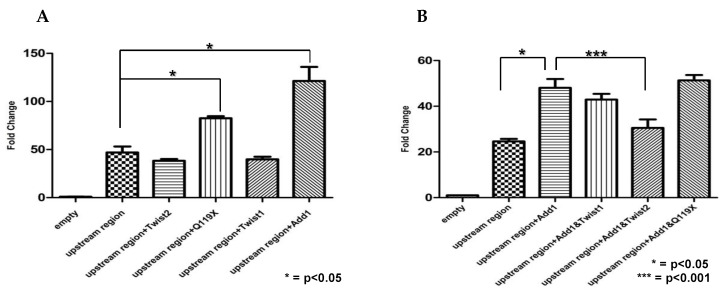
Results: EMSAs showed specific binding of TWIST1 and TWIST2 homodimers, as well as heterodimers with E12, to the more distal E-boxes. An adjoining E-box was bound by ADD1/SREBP1c. EMSA analysis suggested that TWIST2 and ADD1/SREBP1c could compete for binding. Luciferase (luc) reporter assays revealed that the CHRDL1 gene upstream region drives its expression and ADD1/SREBP1c increased it 2.6 times over basal levels. TWIST2, but not the TWIST2-Q119X mutant, blocked activation by ADD1/SREBP1c, but overexpression of TWIST2-Q119X increased luc gene expression. In addition, EMSA competition assays showed that TWIST2, but not TWIST1, competes with ADD1/SREBP1c for DNA binding to the same site.
Conclusions: Formation of an inactive complex between the TWIST2 Q119X and Q65X mutant proteins and ADD1/SREBP1c may prevent repressor binding and allow the binding of other regulators to activate CHRDL1 gene expression.
Keywords: ADD1; BMP signaling; CHRDL1; SREBP1c; Setleis syndrome; TWIST transcription factors; bHLH.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures












References
Grants and funding
LinkOut - more resources
Full Text Sources
