

Advances in the Cystic Fibrosis Drug Development Pipeline
- PMID: 37763239
- PMCID: PMC10532558
- DOI: 10.3390/life13091835
Advances in the Cystic Fibrosis Drug Development Pipeline
Abstract
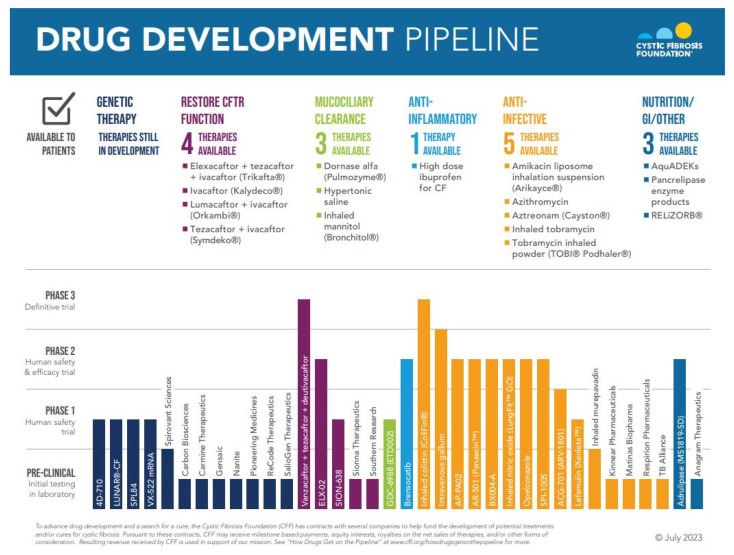
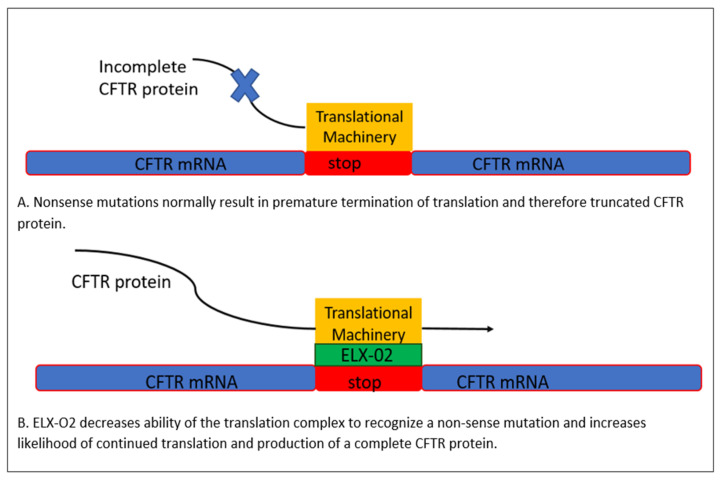
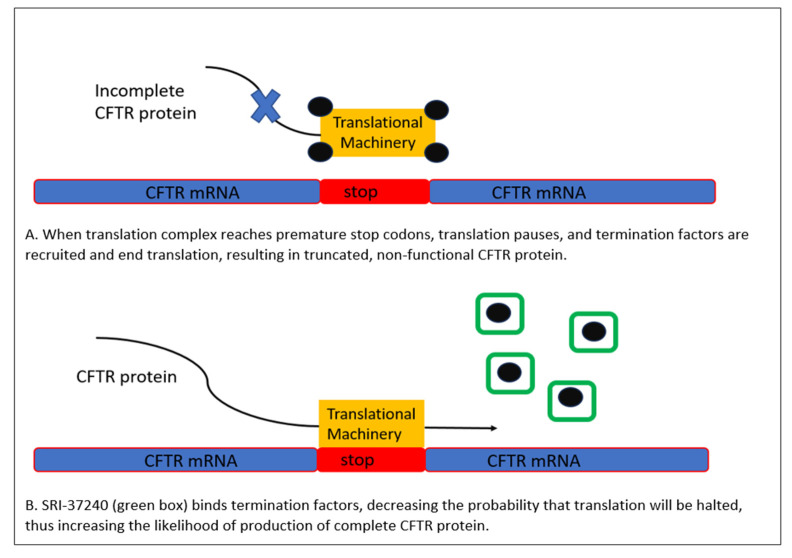
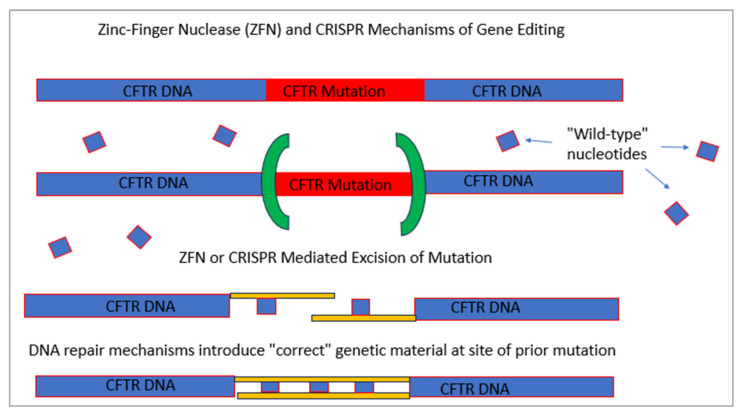
Cystic fibrosis is a genetic disease that results in progressive multi-organ manifestations with predominance in the respiratory and gastrointestinal systems. The significant morbidity and mortality seen in the CF population has been the driving force urging the CF research community to further advance treatments to slow disease progression and, in turn, prolong life expectancy. Enormous strides in medical advancements have translated to improvement in quality of life, symptom burden, and survival; however, there is still no cure. This review discusses the most current mainstay treatments and anticipated therapeutics in the CF drug development pipeline within the mechanisms of mucociliary clearance, anti-inflammatory and anti-infective therapies, restoration of the cystic fibrosis transmembrane conductance regulator (CFTR) protein (also known as highly effective modulator therapy (HEMT)), and genetic therapies. Ribonucleic acid (RNA) therapy, gene transfer, and gene editing are being explored in the hopes of developing a treatment and potential cure for people with CF, particularly for those not responsive to HEMT.
Keywords: anti-inflammatory; antibiotics; bacteriophage; cystic fibrosis (CF); cystic fibrosis transmembrane conductance regulator (CFTR); gene-based therapy; highly effective modulator therapy (HEMT); mucolytic therapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Clancy J.P., Cotton C.U., Donaldson S.H., Solomon G.M., VanDevanter D.R., Boyle M.P., Gentzsch M., Nick J.A., Illek B., Wallenburg J.C., et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019;18:22–34. doi: 10.1016/j.jcf.2018.05.004. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
