

Neurologic orphan diseases: Emerging innovations and role for genetic treatments
- PMID: 37767543
- PMCID: PMC10520757
- DOI: 10.5493/wjem.v13.i4.59
Neurologic orphan diseases: Emerging innovations and role for genetic treatments
Abstract
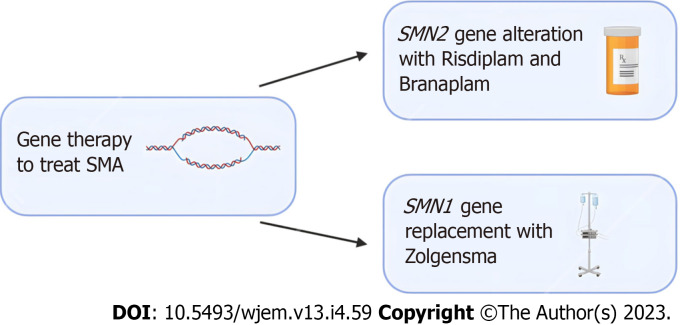
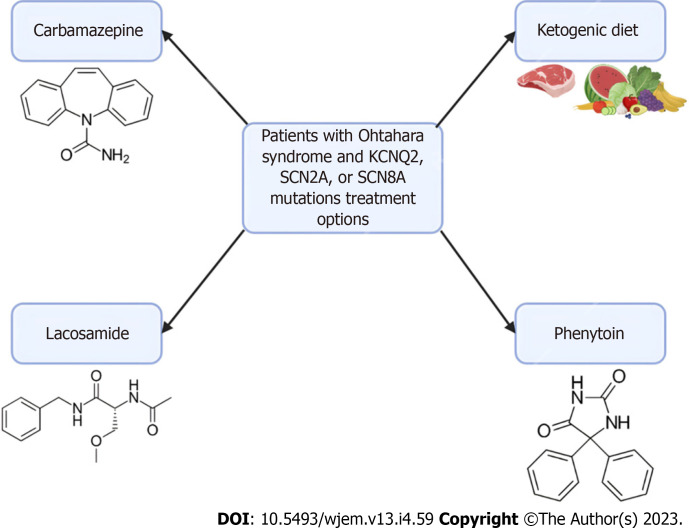
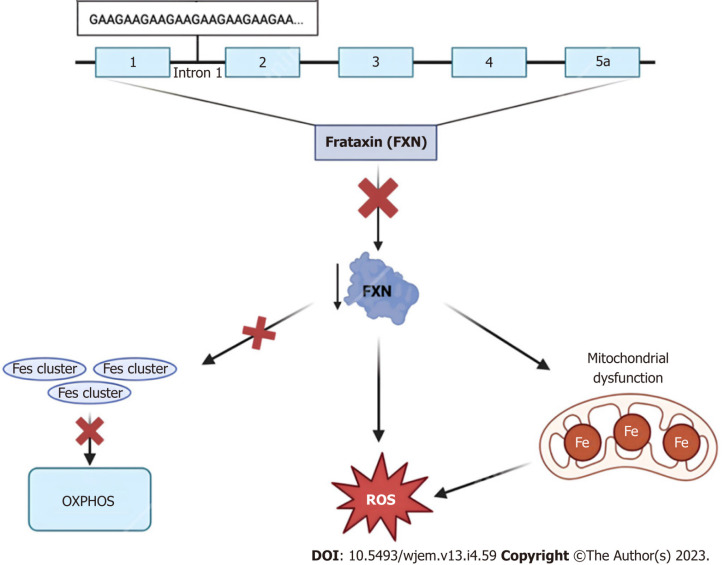
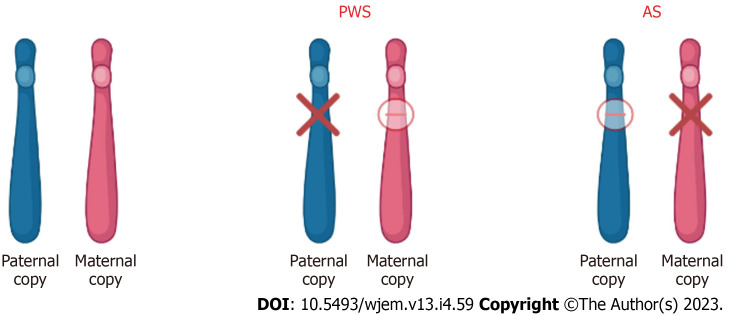
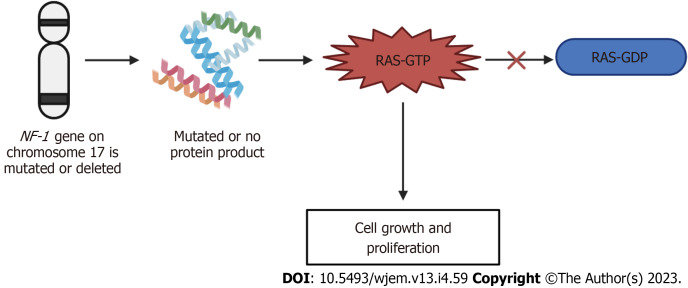
Orphan diseases are rare diseases that affect less than 200000 individuals within the United States. Most orphan diseases are of neurologic and genetic origin. With the current advances in technology, more funding has been devoted to developing therapeutic agents for patients with these conditions. In our review, we highlight emerging options for patients with neurologic orphan diseases, specifically including diseases resulting in muscular deterioration, epilepsy, seizures, neurodegenerative movement disorders, inhibited cognitive development, neuron deterioration, and tumors. After extensive literature review, gene therapy offers a promising route for the treatment of neurologic orphan diseases. The use of clustered regularly interspaced palindromic repeats/Cas9 has demonstrated positive results in experiments investigating its role in several diseases. Additionally, the use of adeno-associated viral vectors has shown improvement in survival, motor function, and developmental milestones, while also demonstrating reversal of sensory ataxia and cardiomyopathy in Friedreich ataxia patients. Antisense oligonucleotides have also been used in some neurologic orphan diseases with positive outcomes. Mammalian target of rapamycin inhibitors are currently being investigated and have reduced abnormal cell growth, proliferation, and angiogenesis. Emerging innovations and the role of genetic treatments open a new window of opportunity for the treatment of neurologic orphan diseases.
Keywords: Adeno-associated virus; Antisense oligonucleotides; Clustered regularly interspaced palindromic repeats/Cas9; Gene therapy; Neurologic orphan diseases; mTOR inhibitors.
©The Author(s) 2023. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: All the authors declare no conflict of interest.
Figures
References
-
- The Lancet Neurology. Rare diseases: maintaining momentum. Lancet Neurol. 2022;21:203. - PubMed
-
- Murphy SM, Puwanant A, Griggs RC Consortium for Clinical Investigations of Neurological Channelopathies (CINCH) and Inherited Neuropathies Consortium (INC) Consortia of the Rare Disease Clinical Research Network. Unintended effects of orphan product designation for rare neurological diseases. Ann Neurol. 2012;72:481–490. - PMC - PubMed
-
- Dowden H, Munro J. Trends in clinical success rates and therapeutic focus. Nat Rev Drug Discov. 2019;18:495–496. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
