

Mixed-Reference Spin-Flip Time-Dependent Density Functional Theory: Multireference Advantages with the Practicality of Linear Response Theory
- PMID: 37767969
- PMCID: PMC10561896
- DOI: 10.1021/acs.jpclett.3c02296
Mixed-Reference Spin-Flip Time-Dependent Density Functional Theory: Multireference Advantages with the Practicality of Linear Response Theory
Abstract
The density functional theory (DFT) and linear response (LR) time-dependent (TD)-DFT are of the utmost importance for routine computations. However, the single reference formulation of DFT suffers in the description of open-shell singlet systems such as diradicals and bond-breaking. LR-TDDFT, on the other hand, finds difficulties in the modeling of conical intersections, doubly excited states, and core-level excitations. In this Perspective, we demonstrate that many of these limitations can be overcome by recently developed mixed-reference (MR) spin-flip (SF)-TDDFT, providing an alternative yet accurate route for such challenging situations. Empowered by the practicality of the LR formalism, it is anticipated that MRSF-TDDFT can become one of the major workhorses for general routine tasks.
Conflict of interest statement
The authors declare no competing financial interest.
Figures






 , where ΔR’s
are displacements with respect to the S0 equilibrium geometry.
For all other electronic structure methods, the MRSF-TDDFT MEP geometries
are utilized by employing a 6-31G* basis set with Cs symmetry restriction.
Adopted with permission from ref (84). Copyright 2009 American Chemical.
, where ΔR’s
are displacements with respect to the S0 equilibrium geometry.
For all other electronic structure methods, the MRSF-TDDFT MEP geometries
are utilized by employing a 6-31G* basis set with Cs symmetry restriction.
Adopted with permission from ref (84). Copyright 2009 American Chemical.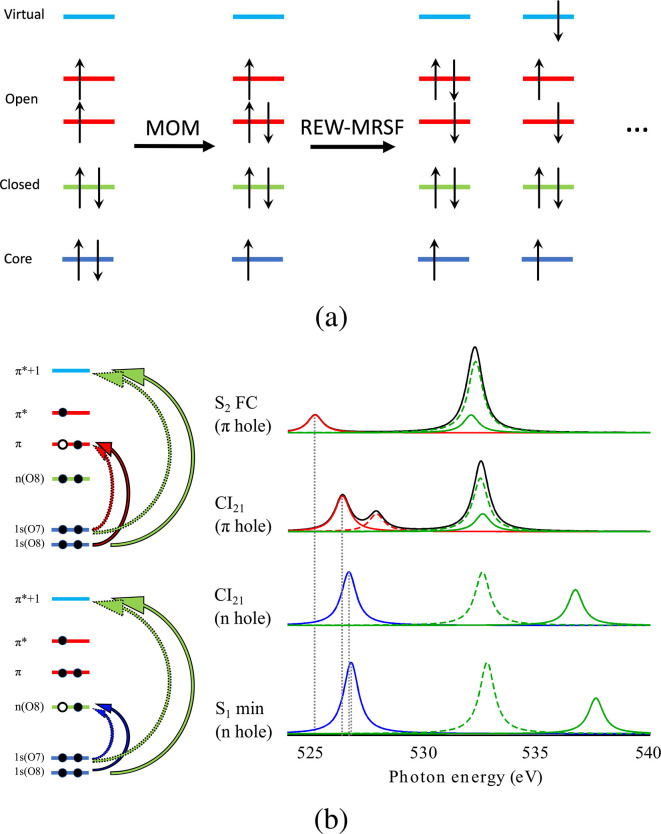

References
-
- Kohn W.; Sham L. J. Self-consistent equations including exchange and correlation effects. Physical review 1965, 140, A1133. 10.1103/PhysRev.140.A1133. - DOI
-
- Squires R. R.; Cramer C. J. Electronic interactions in aryne biradicals. Ab initio calculations of the structures, thermochemical properties, and singlet- triplet splittings of the didehydronaphthalenes. J. Phys. Chem. A 1998, 102, 9072–9081. 10.1021/jp983449b. - DOI
-
- Lee S.; Horbatenko Y.; Filatov M.; Choi C. H. Fast and Accurate Computation of Nonadiabatic Coupling Matrix Elements Using the Truncated Leibniz Formula and Mixed-Reference Spin-Flip Time-Dependent Density Functional Theory. J. Phys. Chem. Lett. 2021, 12, 4722–4728. 10.1021/acs.jpclett.1c00932. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
