

PLCG2-associated immune dysregulation (PLAID) comprises broad and distinct clinical presentations related to functional classes of genetic variants
- PMID: 37769878
- PMCID: PMC11337301
- DOI: 10.1016/j.jaci.2023.08.036
PLCG2-associated immune dysregulation (PLAID) comprises broad and distinct clinical presentations related to functional classes of genetic variants
Abstract
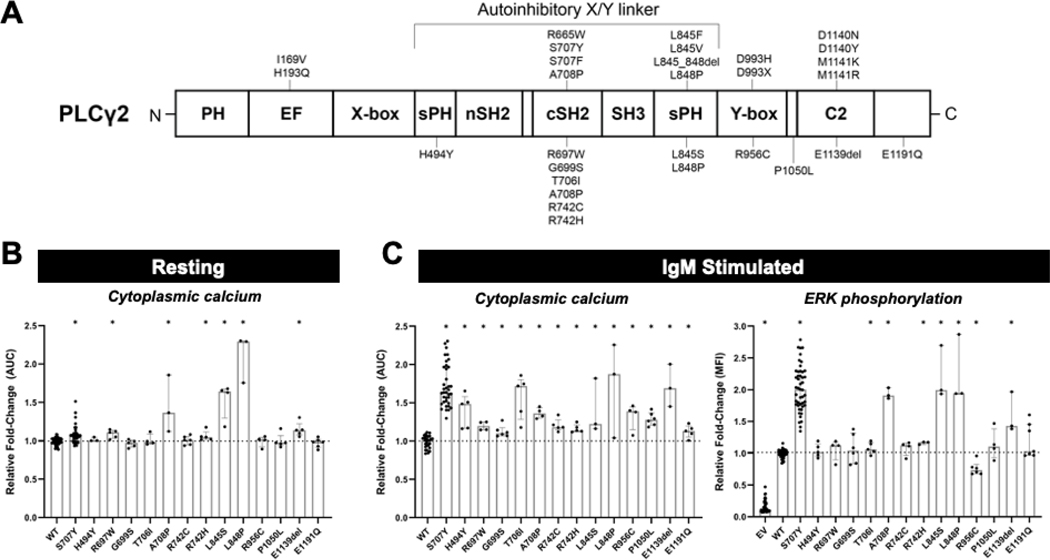
Background: Pathogenic variants of phospholipase C gamma 2 (PLCG2) cause 2 related forms of autosomal-dominant immune dysregulation (ID), PLCγ2-associated antibody deficiency and immune dysregulation (PLAID) and autoinflammatory PLAID (APLAID). Since describing these conditions, many PLCG2 variants of uncertain significance have been identified by clinical sequencing of patients with diverse features of ID.
Objective: We sought to functionally classify PLCG2 variants and explore known and novel genotype-function-phenotype relationships.
Methods: Clinical data from patients with PLCG2 variants were obtained via standardized questionnaire. PLCG2 variants were generated by mutagenesis of enhanced green fluorescent protein (EGFP)-PLCG2 plasmid, which was overexpressed in Plcg2-deficient DT-40 B cells. B-cell receptor-induced calcium flux and extracellular signal-regulated kinase phosphorylation were assayed by flow cytometry. In some cases, stimulation-induced calcium flux was also measured in primary patient cells.
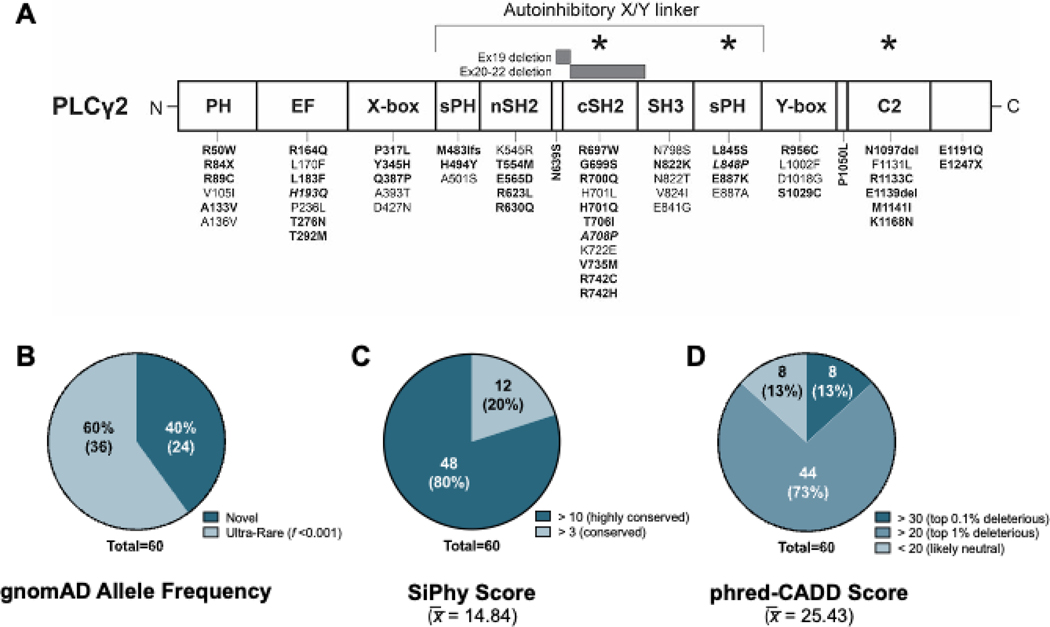
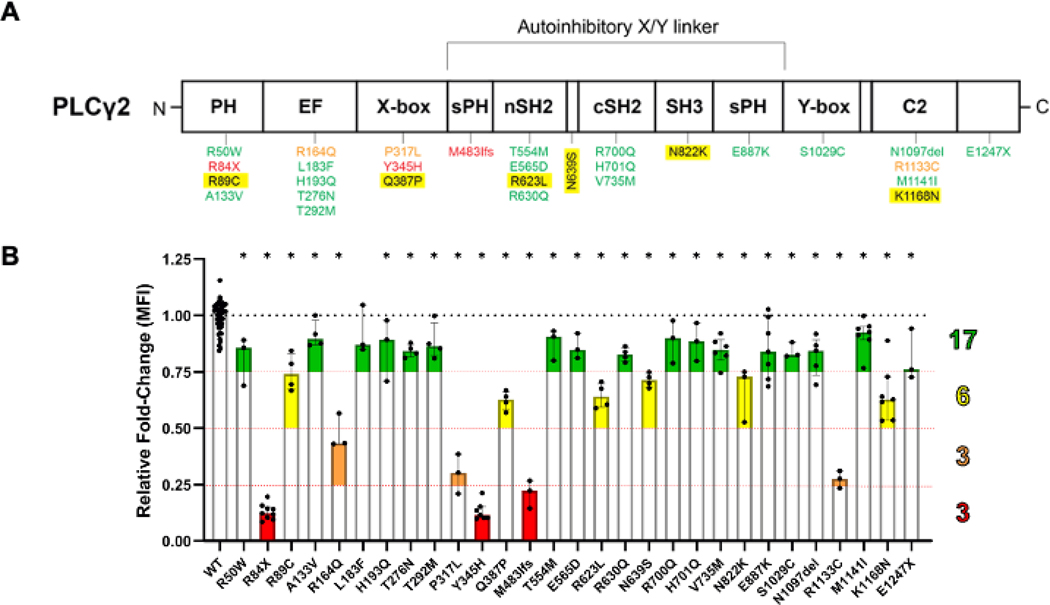
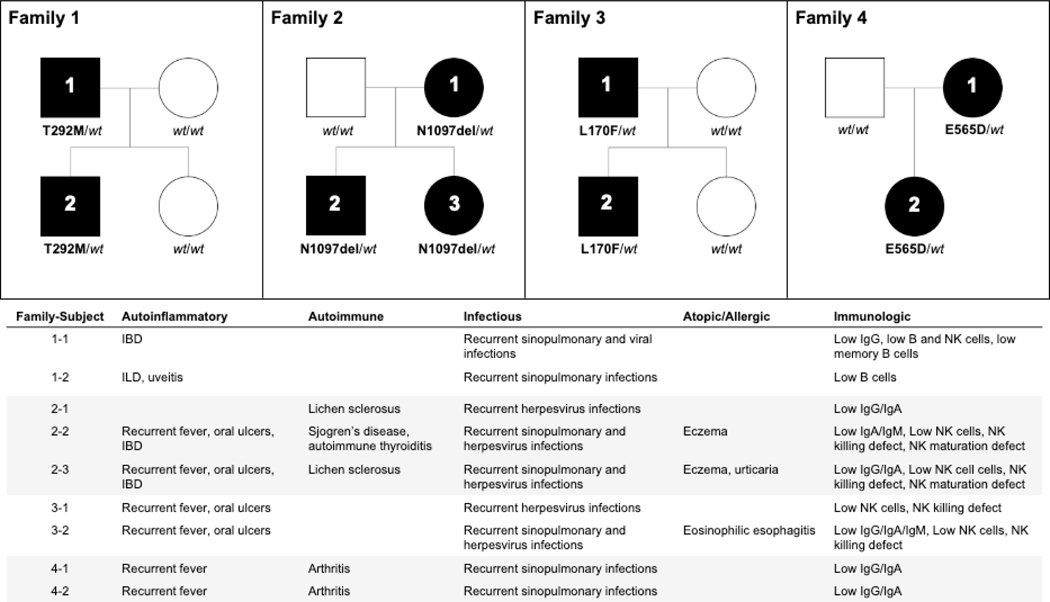
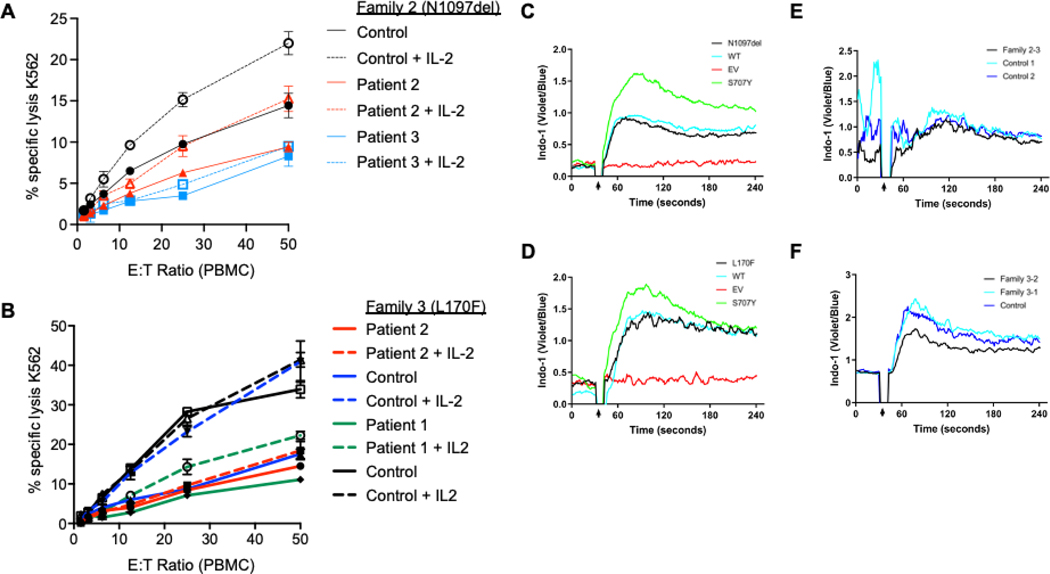
Results: Three-fourths of PLCG2 variants produced functional alteration of B-cell activation, in vitro. Thirteen variants led to gain of function (GOF); however, most functional variants defined a new class of PLCG2 mutation, monoallelic loss of function (LOF). Susceptibility to infection and autoinflammation were common with both GOF and LOF variants, whereas a new phenotypic cluster consisting of humoral immune deficiency, autoinflammation, susceptibility to herpesvirus infection, and natural killer cell dysfunction was observed in association with multiple heterozygous LOF variants detected in both familial and sporadic cases. In some cases, PLCG2 variants produced greater effects in natural killer cells than in B cells.
Conclusions: This work expands the genotypic and phenotypic associations with functional variation in PLCG2, including a novel form of ID in carriers of heterozygous loss of PLCG2 function. It also demonstrates the need for more diverse assays for assessing the impact of PLCG2 variants on human disease.
Keywords: Phospholipase C gamma 2; antibody deficiency; autoinflammation; immune dysregulation; primary immune deficiency; variants of uncertain significance.
Published by Elsevier Inc.
Conflict of interest statement
Disclosure statement This study was funded by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (grant no. ZIA-AR041198 to M.J.O.). Additional support was provided by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (U01-AI122275 to S.H., ZIA-AI000249 to D.M., ZIA-AI001098 to J.D.M., and ZIA-AI001121 to I.S.), and by the Extramural Programs of National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant no. K23-AR070897 to E.S.), National Institute of Allergy and Infectious Diseases (grant no. R01-AI120989 to J.O. and grant no. R01-AI153827 to M.J.B.), National Center for Advancing Translational Sciences (grant no. KL2-TF001862 to J.J.S.), National Institute of Diabetes and Digestive and Kidney Diseases (grant nos. P30DK03485 and RC2DK122532 to S.B.S.), National Human Genome Research Institute (grant no. UM1-HG006542 to Baylor-Hopkins Center for Mendelian Genetics), and National Heart, Lung, and Blood Institute (grant no. R01-HL162642-01A1 to S.C.G.). B.G. is supported by the Deutsche Forschungsgemeinschaft (GR1617/14-1/iPAD; SFB1160/2_B5; RESIST–EXC 2155–Project ID 390874280; the EU-H2020-MSCA-COFUND EURIdoc Programme [no. 101034170]) and the German Federal Ministry of Education and Research (grant no. GAIN 01GM1910A). E.H. is supported by the Bank of Montreal Chair of Pediatric Immunology. F.H. received funding from the Else Kröner-Fresenius Stiftung (grant no. 2017_A110) and the German Federal Ministry of Education and Research (grant no. 01GM1910C). J.H. (Joud Hajjar) was supported by the Immune Deficiency Foundation, the US immunodeficiency network, Chao-physician Scientist award, the Texas Medical Center Digestive Diseases Center, and the Jeffrey Modell Foundation. E.J. is supported by the Association Maladie Foie Enfants (Malakoff, France), Association “Pour Louis 1000 Foie Merci” (Fournet-Luisans, France), and Fondation Rumsey-Cartier (Genève, Switzerland). I.M. is supported by the Research Foundation (grant nos. G0B5120N and G0E8420N) and the Jeffrey Modell Foundation. I.M. has received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 948959). M.R.J.S. received funding from the Helsinki University Hospital Funds, the Jane and Aatos Erkko Foundation, and the Finnish Foundation for Pediatric Research. S.B.S. is supported by the Helmsley Charitable Trust, the Woplow Family Chair in IBD Treatment and Research, the Translational Investigator Service at Boston Children’s Hospital, and the Children’s Rare Disease Cohort Study at Boston Children’s Hospital. J.E.W. is supported by the Jeffrey Modell Foundation and Robert A. Good Endowment, University of South Florida. Disclosure of potential conflict of interest: I.I.C. is on the advisory board of Enzyvant; is a consultant for Pharming; and is a medical writer for UpToDate. K.C. is on the advisory board of Takeda and the Speaker’s Bureau for Horizon Pharma. S.C.G. is a consultant for Janssen. A.A.G. is a coinvestigator in studies with Innate Pharma, CRISPR Therapeutics, StemLine Therapeutics, and Kyowa Kyrin; and is a consultant for StemLine Therapeutics and BluePrints Medicine. E.H. is an advisor to Jasper Therapeutics, Octapharma, CSL Behring, and Takeda; and is a consultant for Vivet Therapeutics and laboratoire CTRS, France. I.M. is on the advisory board of Boehringer Ingelheim; and receives research funding (paid to UZ Leuven) from CSL Behring. S.B.S. was on the scientific advisory board for Pfizer, Pandion, Celgene, Eli Lilly, Takeda, Cosmo Pharmaceuticals, Merck, Sonoma Biotherapeutics, and EcoR1; received grant support from Pfizer, Amgen-Takeda, and Novartis; and is a consultant for Amgen, Kyverna, BMS, Merck, Third Rock, 89bio, GentiBio, and Apple Tree Life Sciences. T.T. is a consultant for Takeda, Horizon, X4 Pharmaceuticals, and Pharming Healthcare; serves on the Data Safety and Monitoring Board for Takeda (formerly Baxalta); and receives research funding from the Paul G. Allen Family Foundation, NIH, and Eli Lilly. J.E.W. receives grant/research/clinical trial support from Takeda, Janssen, Chiesi, MustangBio, ADMA Biologicals, Octapharma, X4 Pharmaceuticals, Novartis, Regeneron, and Bristol-Myers Squibb; is a consultant or advisory board member for Takeda, X4 Pharmaceuticals, CSL Behring, Grifols, ADMA Biologicals, Enzyvant, and Regeneron; is on the Speaker’s Bureau for Takeda; and is a medical writer for UpToDate. B.R.W. receives research support from Merck and Swedish Orphan Biovitrum; receives clinical trial support from AstraZeneca and Blueprint Medicines; receives speaker’s fees from Takeda; and serves on the Data Safety and Monitoring Board for REDHART2 clinical trial. The rest of the authors declare that they have no relevant conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- ZIA AR041198/ImNIH/Intramural NIH HHS/United States
- ZIA AI001121/ImNIH/Intramural NIH HHS/United States
- U54 HG006542/HG/NHGRI NIH HHS/United States
- R01 AI120989/AI/NIAID NIH HHS/United States
- R01 HL162642/HL/NHLBI NIH HHS/United States
- ZIA AI001098/ImNIH/Intramural NIH HHS/United States
- UM1 HG006542/HG/NHGRI NIH HHS/United States
- K23 AR070897/AR/NIAMS NIH HHS/United States
- ZIA AI000249/ImNIH/Intramural NIH HHS/United States
- U01 AI122275/AI/NIAID NIH HHS/United States
- RC2 DK122532/DK/NIDDK NIH HHS/United States
- R01 AI153827/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
