

Integrated molecular and multiparametric MRI mapping of high-grade glioma identifies regional biologic signatures
- PMID: 37770427
- PMCID: PMC10539500
- DOI: 10.1038/s41467-023-41559-1
Integrated molecular and multiparametric MRI mapping of high-grade glioma identifies regional biologic signatures
Abstract
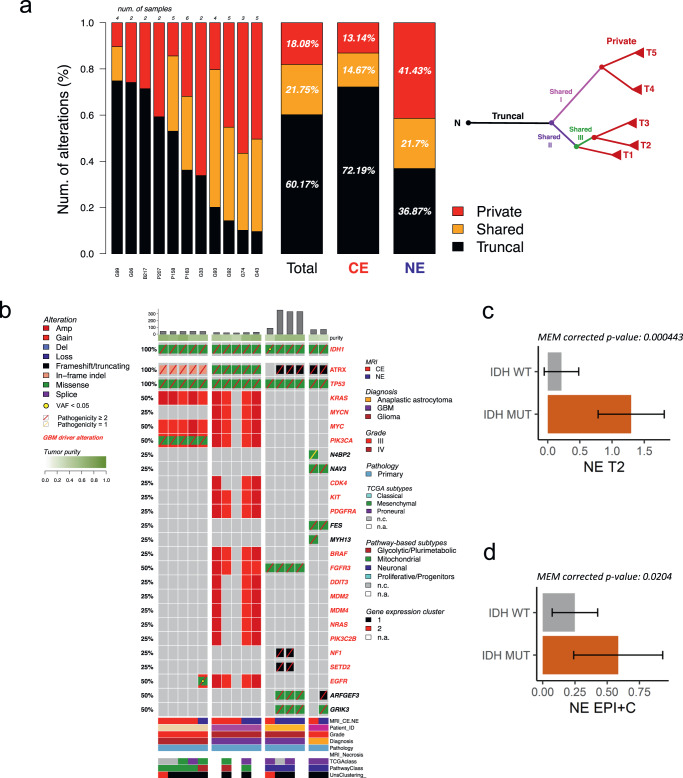
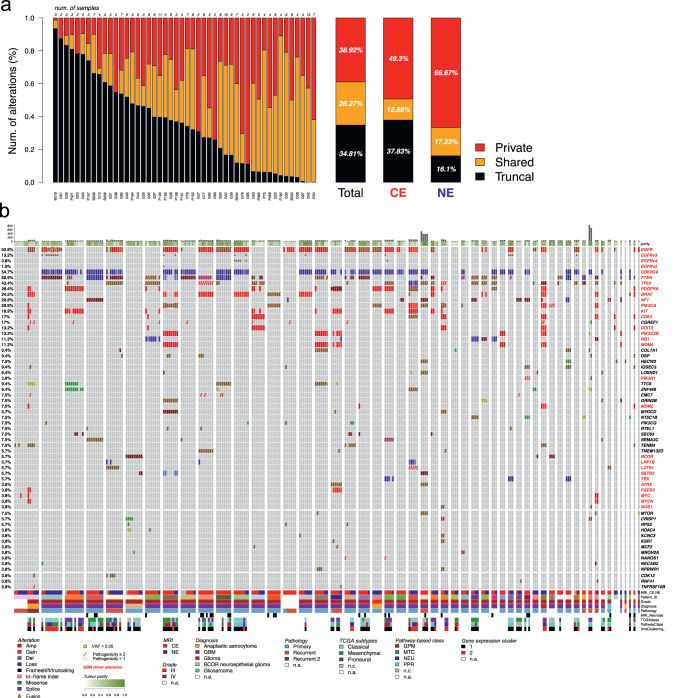
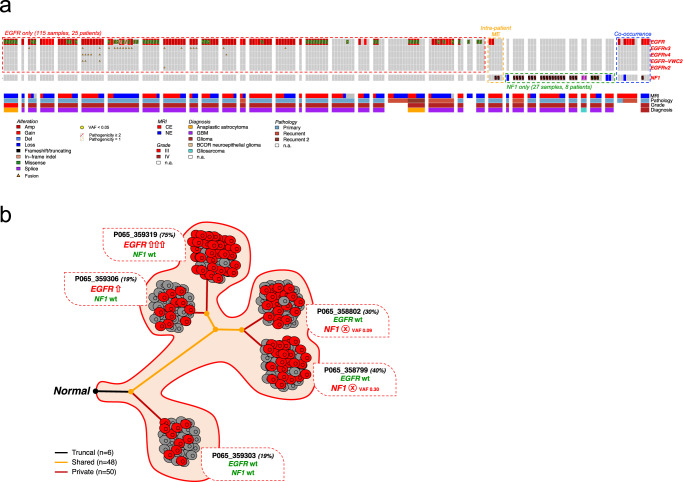
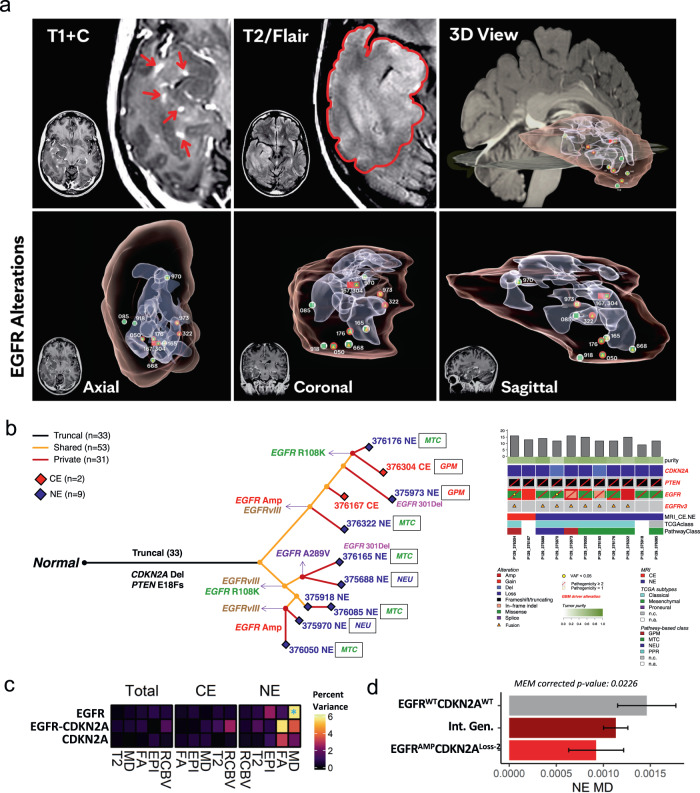
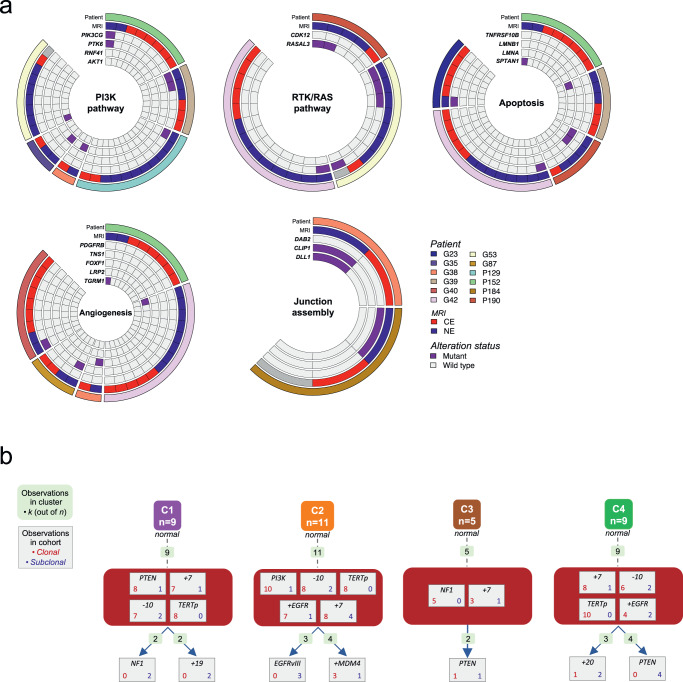
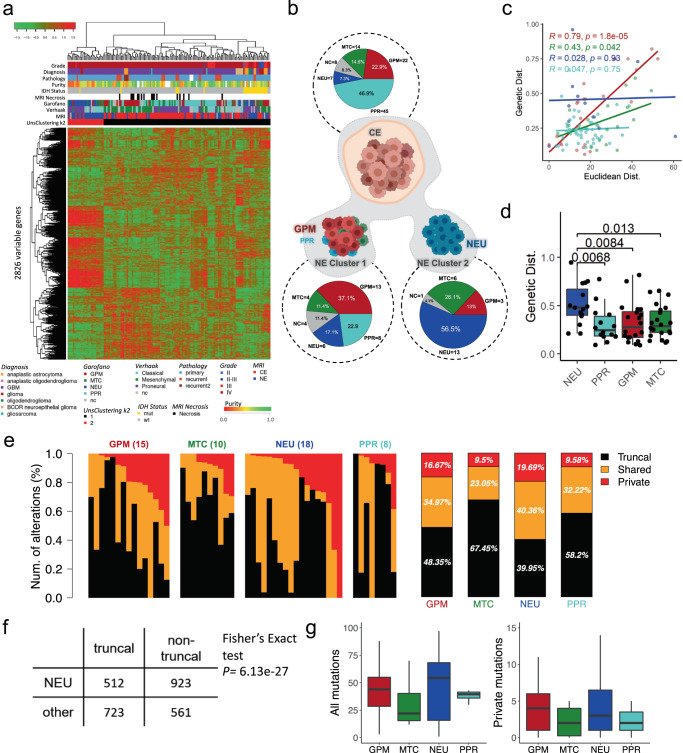
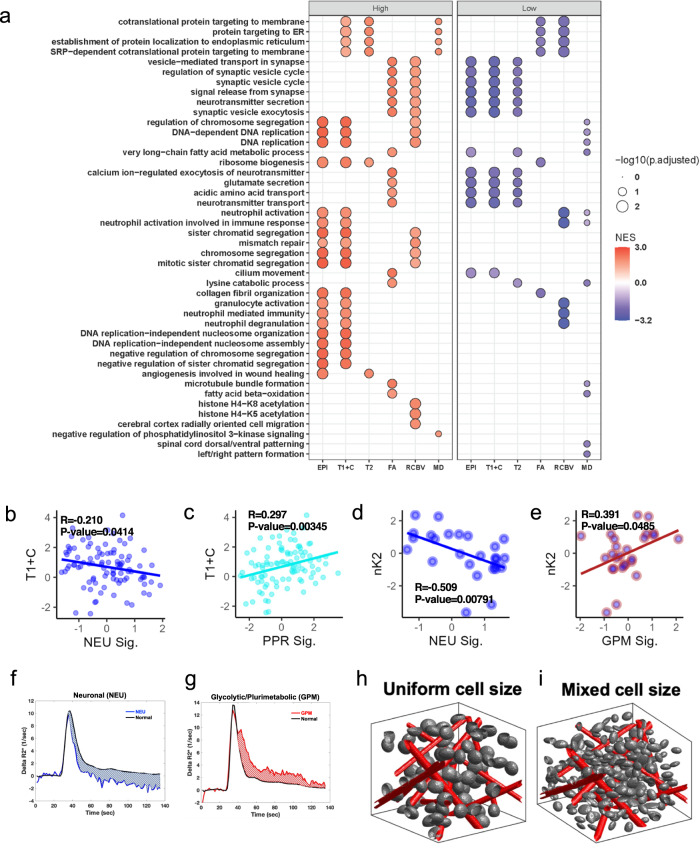
Sampling restrictions have hindered the comprehensive study of invasive non-enhancing (NE) high-grade glioma (HGG) cell populations driving tumor progression. Here, we present an integrated multi-omic analysis of spatially matched molecular and multi-parametric magnetic resonance imaging (MRI) profiling across 313 multi-regional tumor biopsies, including 111 from the NE, across 68 HGG patients. Whole exome and RNA sequencing uncover unique genomic alterations to unresectable invasive NE tumor, including subclonal events, which inform genomic models predictive of geographic evolution. Infiltrative NE tumor is alternatively enriched with tumor cells exhibiting neuronal or glycolytic/plurimetabolic cellular states, two principal transcriptomic pathway-based glioma subtypes, which respectively demonstrate abundant private mutations or enrichment in immune cell signatures. These NE phenotypes are non-invasively identified through normalized K2 imaging signatures, which discern cell size heterogeneity on dynamic susceptibility contrast (DSC)-MRI. NE tumor populations predicted to display increased cellular proliferation by mean diffusivity (MD) MRI metrics are uniquely associated with EGFR amplification and CDKN2A homozygous deletion. The biophysical mapping of infiltrative HGG potentially enables the clinical recognition of tumor subpopulations with aggressive molecular signatures driving tumor progression, thereby informing precision medicine targeting.
© 2023. Springer Nature Limited.
Conflict of interest statement
Precision Oncology Insights (co-founders: K.R.S., L.S.H.); Imaging Biometrics (medical advisory board L.S.H., employee T.D., consultant T.R.J.); IQ-AI Ownership Interest (K.M.S., T.D., L.S.H.), Prism Clinical Imaging Ownership Interest (K.M.S.), Imaging Biometrics Financial Interest (K.M.S., T.D., L.S.H.); The International Genomics Consortium (scientific advisor: N.L.T.), the remaining authors have no conflicts of interest to disclose.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
