

Efficient gene orthology inference via large-scale rearrangements
- PMID: 37770945
- PMCID: PMC10540461
- DOI: 10.1186/s13015-023-00238-y
Efficient gene orthology inference via large-scale rearrangements
Abstract
Background: Recently we developed a gene orthology inference tool based on genome rearrangements (Journal of Bioinformatics and Computational Biology 19:6, 2021). Given a set of genomes our method first computes all pairwise gene similarities. Then it runs pairwise ILP comparisons to compute optimal gene matchings, which minimize, by taking the similarities into account, the weighted rearrangement distance between the analyzed genomes (a problem that is NP-hard). The gene matchings are then integrated into gene families in the final step. The mentioned ILP includes an optimal capping that connects each end of a linear segment of one genome to an end of a linear segment in the other genome, producing an exponential increase of the search space.
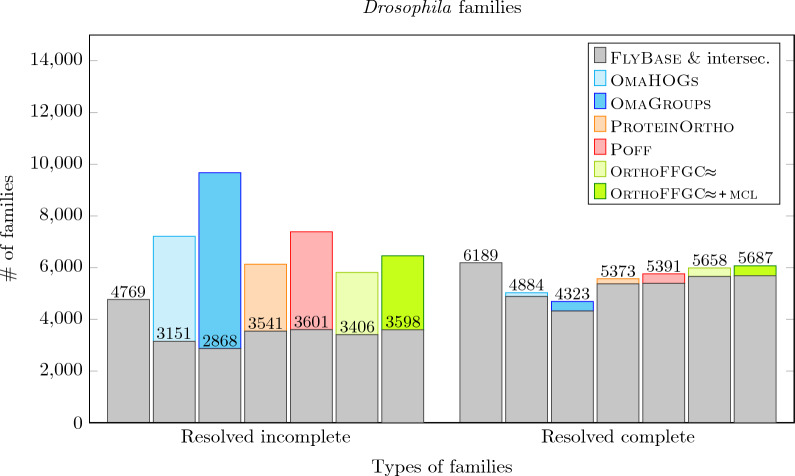
Results: In this work, we design and implement a heuristic capping algorithm that replaces the optimal capping by clustering (based on their gene content intersections) the linear segments into [Formula: see text] subsets, whose ends are capped independently. Furthermore, in each subset, instead of allowing all possible connections, we let only the ends of content-related segments be connected. Although there is no guarantee that m is much bigger than one, and with the possible side effect of resulting in sub-optimal instead of optimal gene matchings, the heuristic works very well in practice, from both the speed performance and the quality of computed solutions. Our experiments on primate and fruit fly genomes show two positive results. First, for complete assemblies of five primates the version with heuristic capping reports orthologies that are very similar to the orthologies computed by the version of our tool with optimal capping. Second, we were able to efficiently analyze fruit fly genomes with incomplete assemblies distributed in hundreds or even thousands of contigs, obtaining gene families that are very similar to [Formula: see text] families. Indeed, our tool inferred a higher number of complete cliques, with a higher intersection with [Formula: see text], when compared to gene families computed by other inference tools. We added a post-processing for refining, with the aid of the [Formula: see text] algorithm, our ambiguous families (those with more than one gene per genome), improving even more the accuracy of our results. Our approach is implemented into a pipeline incorporating the pre-computation of gene similarities and the post-processing refinement of ambiguous families with [Formula: see text]. Both the original version with optimal capping and the new modified version with heuristic capping can be downloaded, together with their detailed documentations, at https://gitlab.ub.uni-bielefeld.de/gi/FFGC or as a Conda package at https://anaconda.org/bioconda/ffgc .
Keywords: Comparative genomics; Double-cut-and-join; Gene orthology; Indels.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
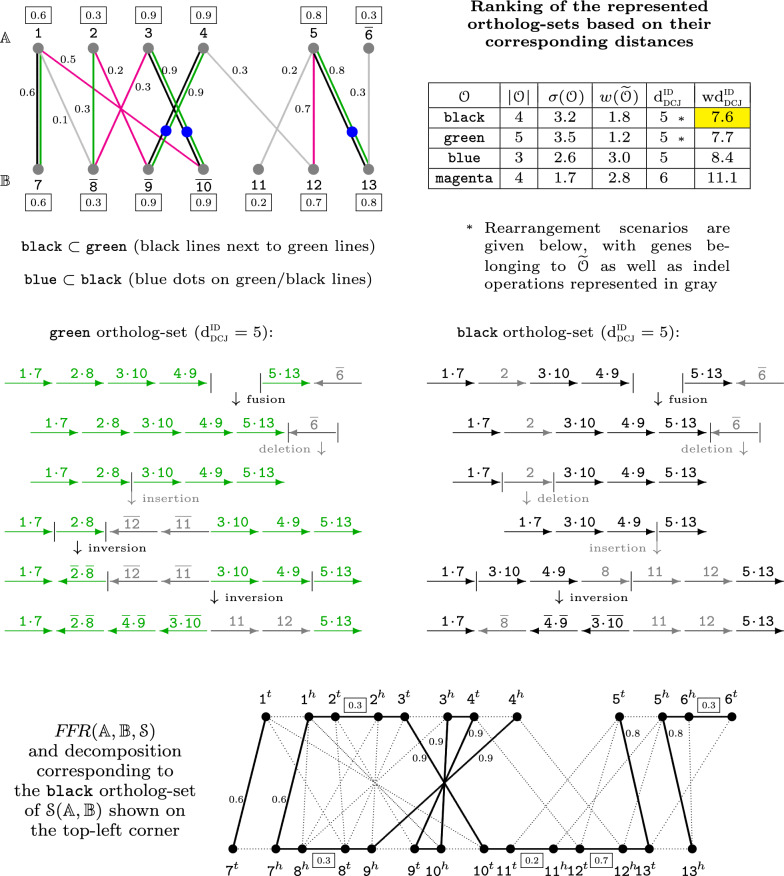
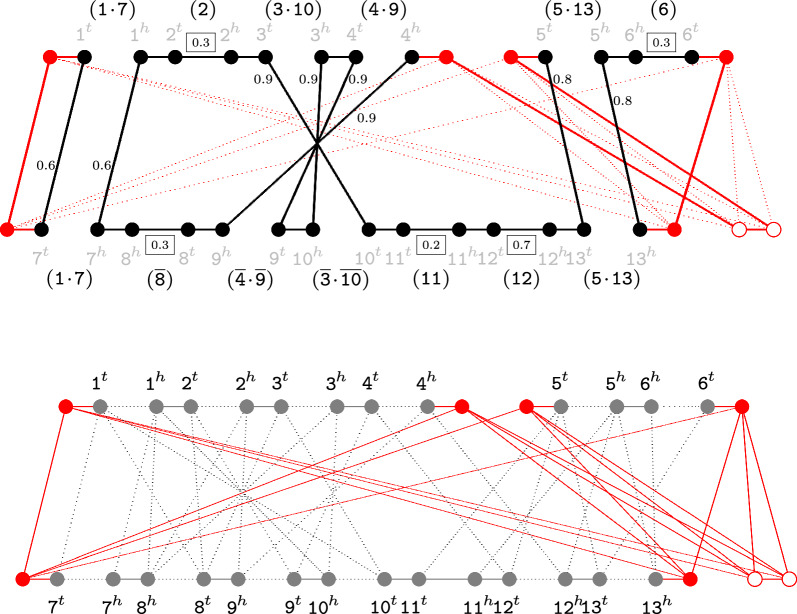
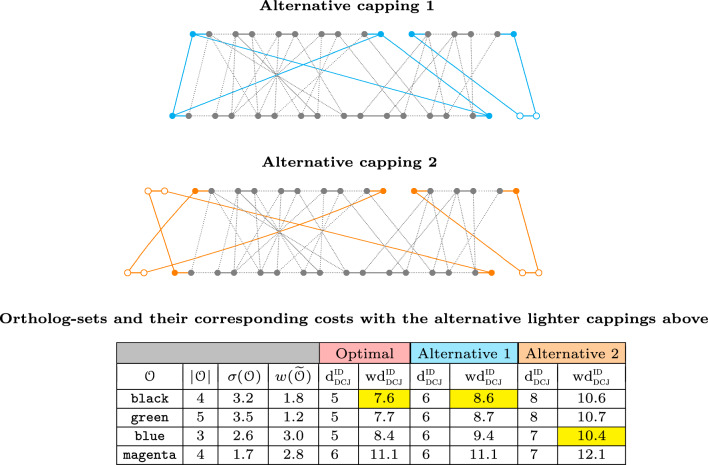


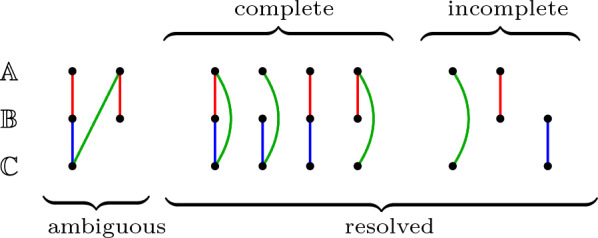

References
-
- Bergeron A, Mixtacki J, Stoye J. A unifying view of genome rearrangements. In: Proc. of WABI. Lecture Notes in Bioinformatics, 2006;4175:163–173.
-
- Hannenhalli S, Pevzner PA. Transforming men into mice (polynomial algorithm for genomic distance problem). In: Proc. of FOCS, 1995:581–592.
-
- Bryant D. The complexity of calculating exemplar distances. In: Sankoff D, Nadeau JH, editors. Comparative Genomics. Computational Biology Series. London: Kluver Academic Publishers; 2000. pp. 207–211.
LinkOut - more resources
Full Text Sources
Miscellaneous
