

Epitope-engineered human hematopoietic stem cells are shielded from CD123-targeted immunotherapy
- PMID: 37773046
- PMCID: PMC10541312
- DOI: 10.1084/jem.20231235
Epitope-engineered human hematopoietic stem cells are shielded from CD123-targeted immunotherapy
Abstract
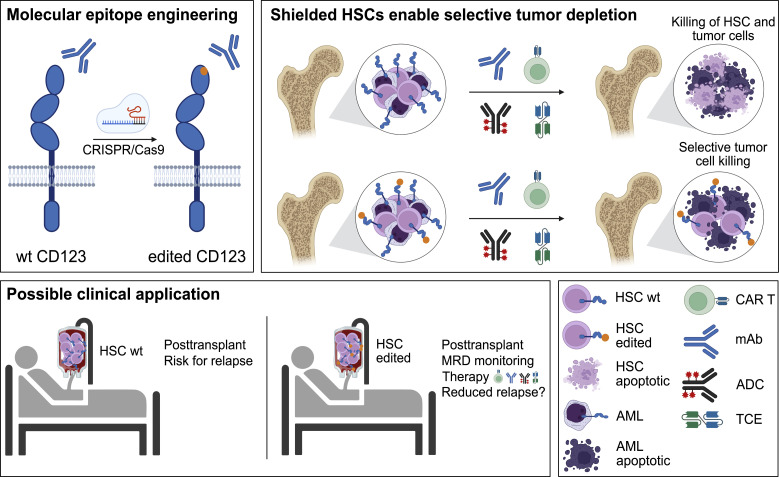
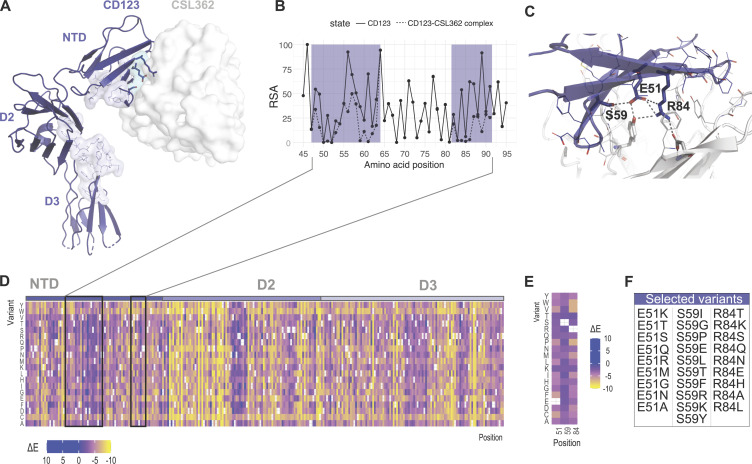
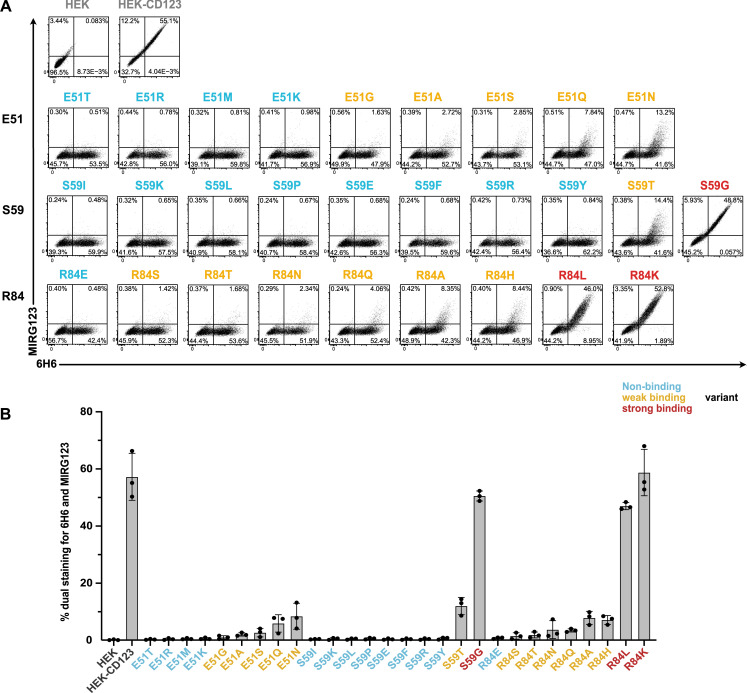
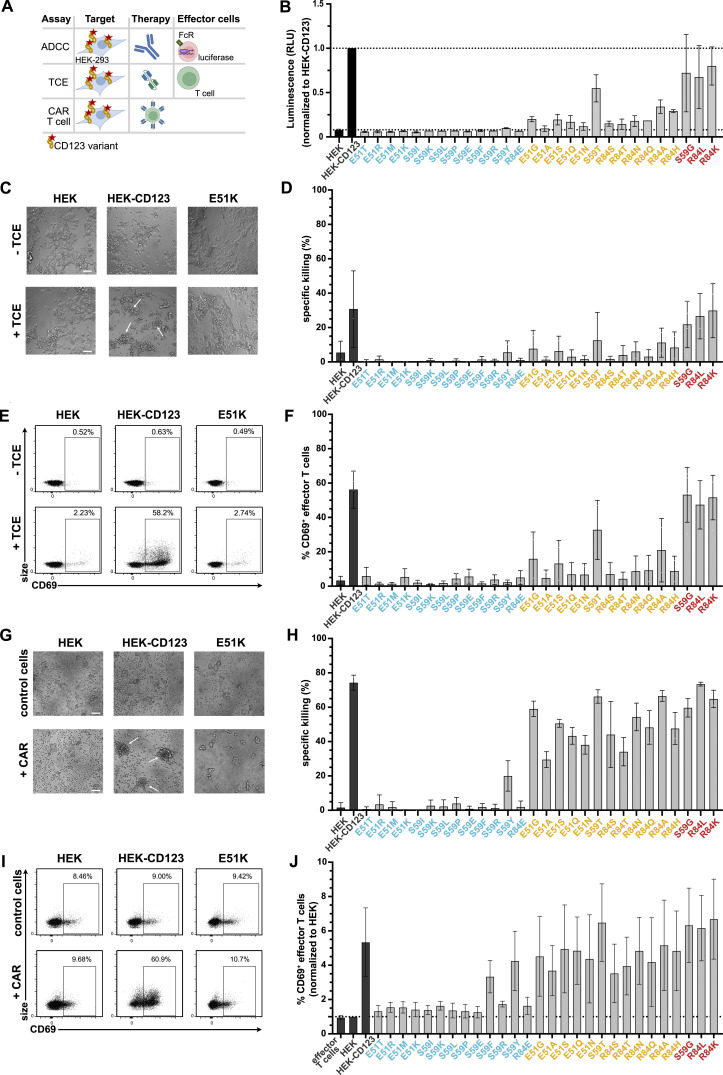
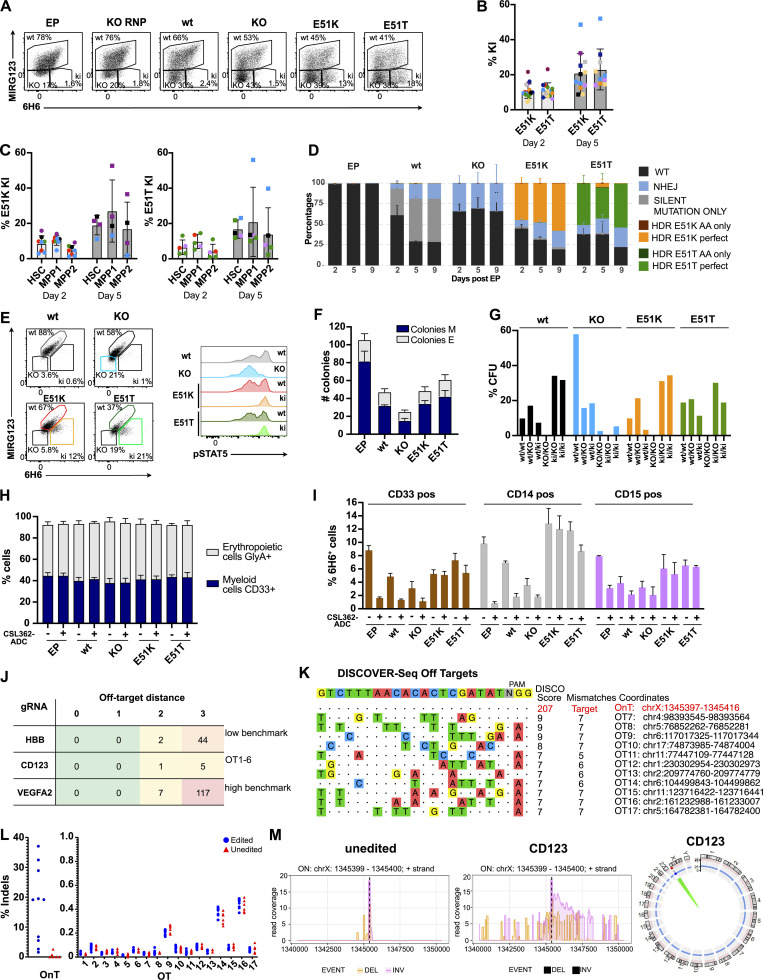
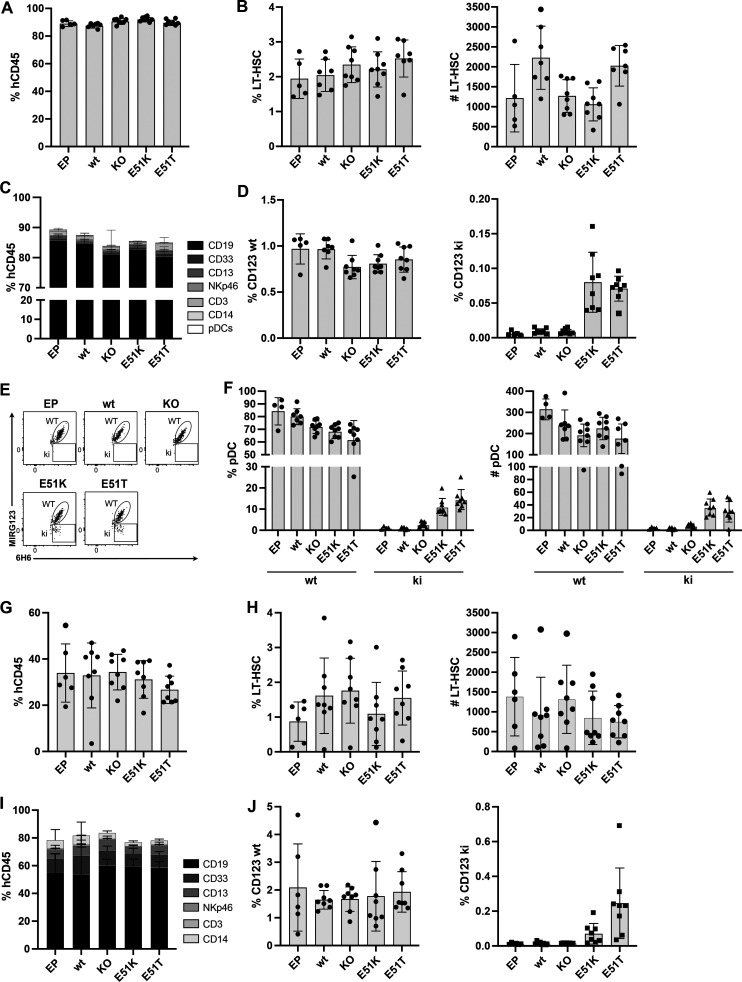
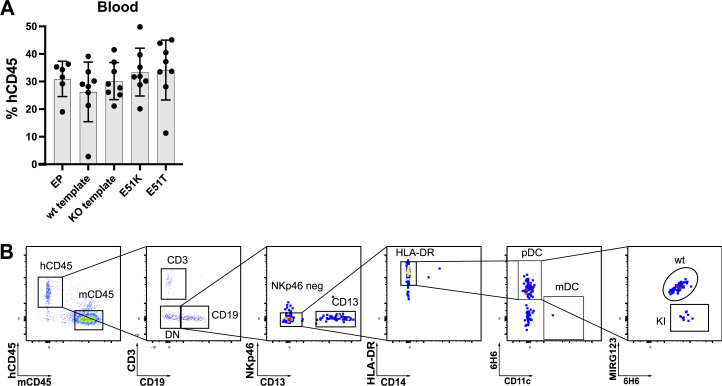
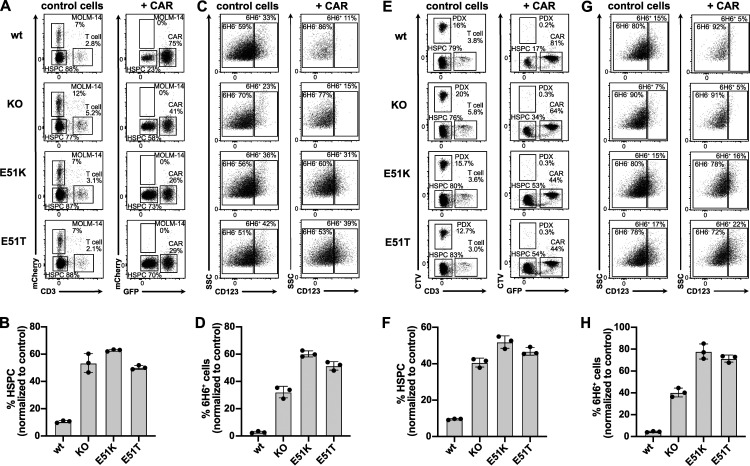
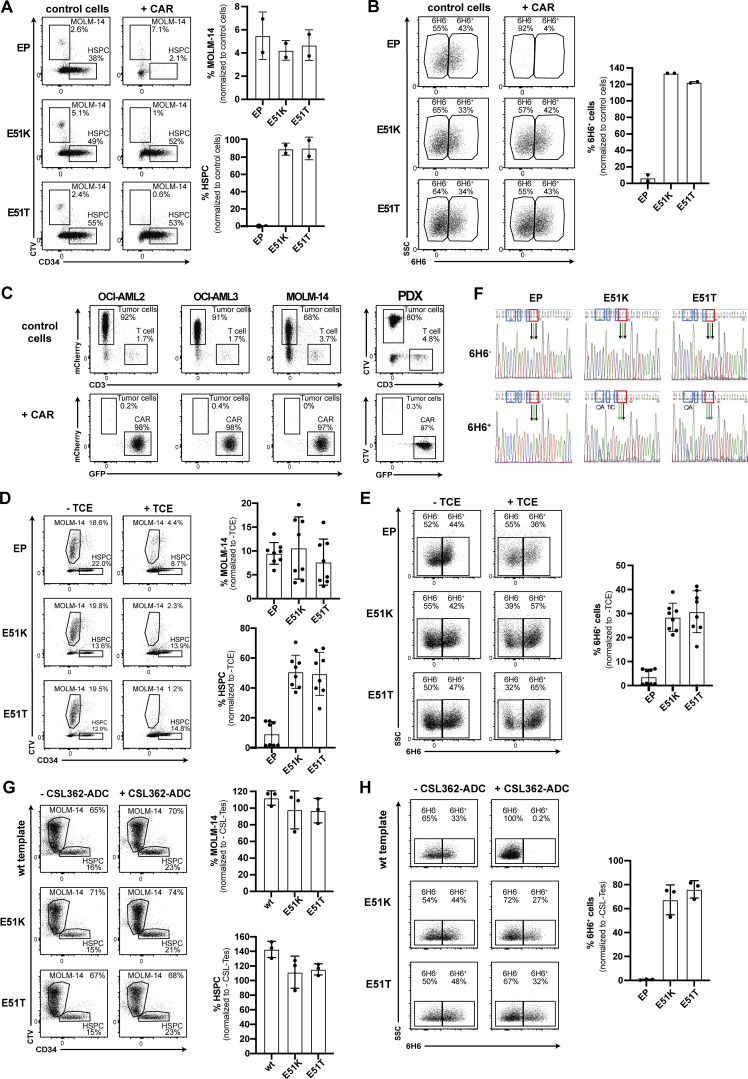
Targeted eradication of transformed or otherwise dysregulated cells using monoclonal antibodies (mAb), antibody-drug conjugates (ADC), T cell engagers (TCE), or chimeric antigen receptor (CAR) cells is very effective for hematologic diseases. Unlike the breakthrough progress achieved for B cell malignancies, there is a pressing need to find suitable antigens for myeloid malignancies. CD123, the interleukin-3 (IL-3) receptor alpha-chain, is highly expressed in various hematological malignancies, including acute myeloid leukemia (AML). However, shared CD123 expression on healthy hematopoietic stem and progenitor cells (HSPCs) bears the risk for myelotoxicity. We demonstrate that epitope-engineered HSPCs were shielded from CD123-targeted immunotherapy but remained functional, while CD123-deficient HSPCs displayed a competitive disadvantage. Transplantation of genome-edited HSPCs could enable tumor-selective targeted immunotherapy while rebuilding a fully functional hematopoietic system. We envision that this approach is broadly applicable to other targets and cells, could render hitherto undruggable targets accessible to immunotherapy, and will allow continued posttransplant therapy, for instance, to treat minimal residual disease (MRD).
© 2023 Marone et al.
Conflict of interest statement
Disclosures: R. Marone reported a patent to immunologically discernible cell variants for use in cell therapy licensed (Cimeio Therapeutics AG) and a patent to discernible cell surface protein variants for use in cell therapy licensed (Cimeio Therapeutics AG). E. Landmann reported a patent to WO2023/012367 licensed (Cimeio Therapeutics AG). A. Devaux reported a patent to WO2023/012367 pending (Cimeio Therapeutics AG). R. Lepore reported personal fees from Cimeio Therapeutics during the conduct of the study; in addition, R. Lepore had a patent to WO2018/083071 licensed (Cimeio Therapeutics AG) and a patent to WO2023/012367 licensed (Cimeio Therapeutics AG). F. Simonetta reported grants from Gilead, Novartis, and BMS/Cellgene; and “other” from Incyte, Janssen, AstraZeneca, and Neovii outside the submitted work. G. Andrieux reported a patent to US11319580B2 licensed. A. Wiederkehr reported “other” from Ridgeline discovery during the conduct of the study; in addition, A. Wiederkehr had a patent to WO2023/012367 pending (Cimeio Therapeutics). A. Sinopoli reported “other” from Ridgeline Discovery during the conduct of the study; in addition, A. Sinopoli had a patent to WO2023/012367 pending (Cimeio Therapeutics). V. Do Sacramento reported “other” from Cimeio Therapeutics during the conduct of the study. A. Haydn reported “other” from Ridgeline Discovery during the conduct of the study; in addition, A. Haydn had a patent to WO2023/012367 pending (Cimeio Therapeutics). L. Garcia-Prat reported “other” from Cimeio Therapeutics during the conduct of the study; in addition, L. Garcia-Prat had a patent to WO2023/012367 pending (Cimeio Therapeutics). A. Camus reported “other” from Cimeio Therapeutics during the conduct of the study; in addition, A. Camus had a patent to WO2023/012367 pending. L. Bordoli reported a patent to WO2018/083071 licensed (Cimeio Therapeutics AG). T. Schwede reported a patent to WO2018/083071 licensed (Cimeio Therapeutics AG). M. Porteus reported grants from Cimeio, “other” from CRISPR Tx, Allogene Tx, and Graphite Bio, and personal fees from Versant Ventures outside the submitted work. J.E. Corn reported grants from Cimeio Therapeutics outside the submitted workand is a cofounder and board member of Spotlight Therapeutics, an SAB member of Mission Therapeutics, Relation Therapeutics, Hornet Bio, and the Joint AstraZeneca-CRUK Functional Genomics Centre, and a consultant for Cimeio Therapeutics. The lab of J.E. Corn has funded collaborations with Allogene and Cimeio. J.E. Corn is supported by the NOMIS Foundation and the Lotte and Adolf Hotz-Sprenger Stiftung. T. Cathomen reported personal fees from Cimeio Therapeutics during the conduct of the study; personal fees from Excision Biotherapeutics, GenCC, and Novo Nordisk, and grants from Cellectis outside the submitted work; in addition, T. Cathomen had a patent to CAST-Seq issued. T.I. Cornu reported grants from Cimeio Therapeutics during the conduct of the study, and grants from Cellectis outside the submitted work. S. Urlinger reported “other” from Cimeio Therapeutics during the conduct of the study; in addition, S. Urlinger had a patent to WO2023/012367 pending (Cimeio Therapeutics). L.T. Jeker reported grants non-financial support from Cimeio Therapeutics AG during the conduct of the study; in addition, L.T. Jeker had a patent to WO2017/186718 licensed (Cimeio Therapeutics AG), a patent to WO2018/083071 licensed (Cimeio Therapeutics AG), and a patent to WO2023/012367 licensed (Cimeio Therapeutics AG); is a co-founder and board member of Cimeio Therapeutics AG, and holding equity in Cimeio Therapeutics AG. No other disclosures were reported.
Figures
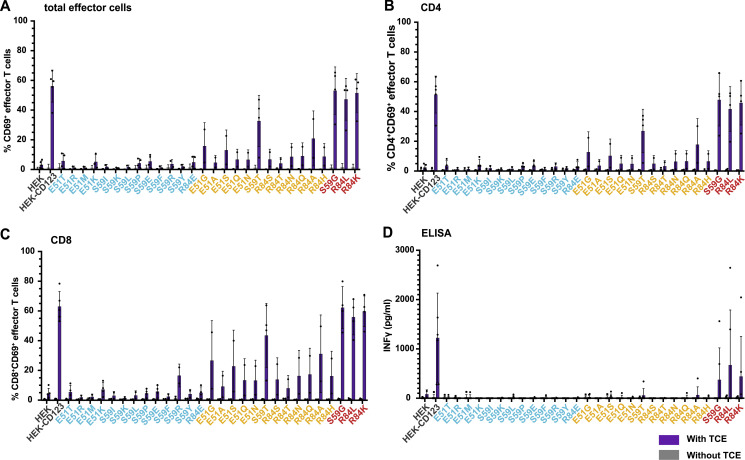
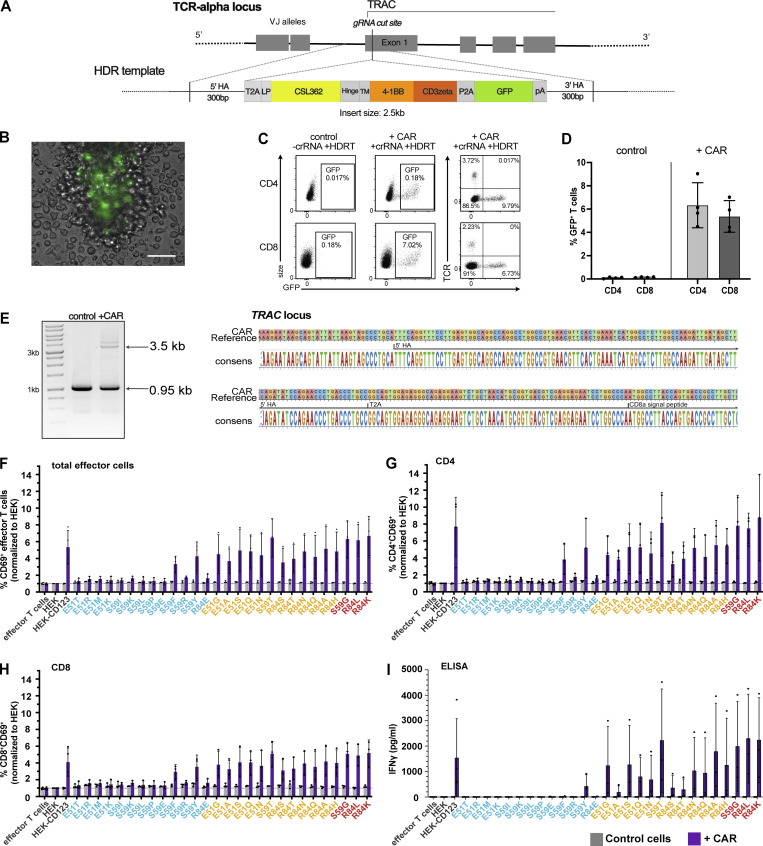
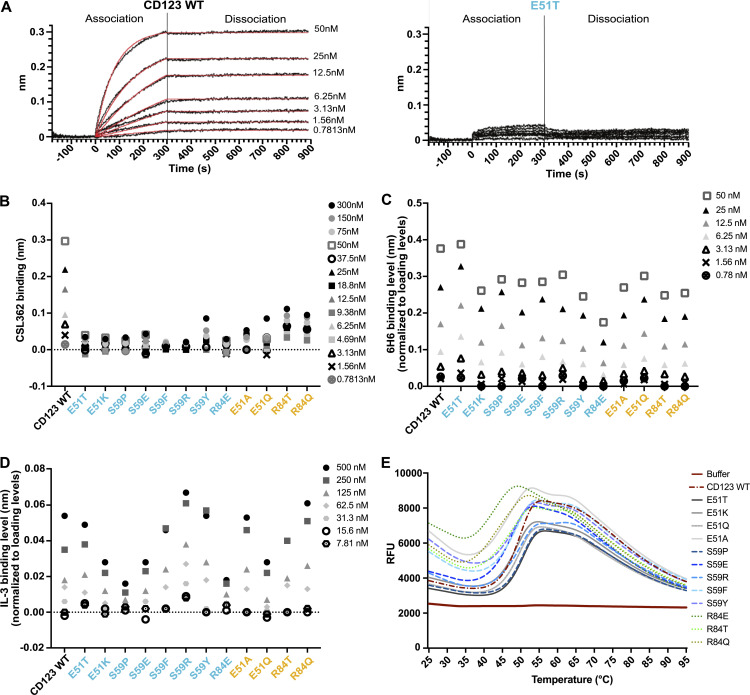
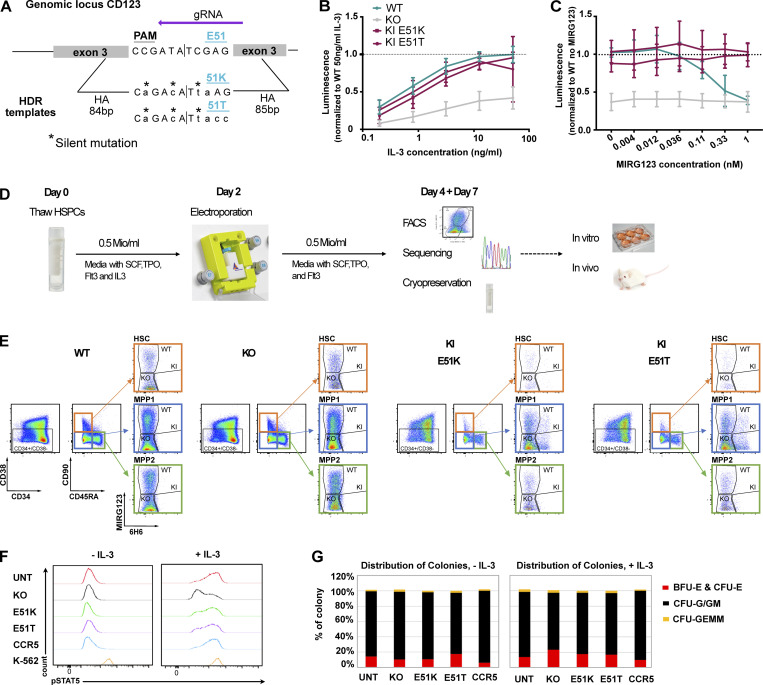
Comment in
-
Twisted: Escape of epitope-edited healthy cells from immune attack.J Exp Med. 2023 Dec 4;220(12):e20231635. doi: 10.1084/jem.20231635. Epub 2023 Oct 11. J Exp Med. 2023. PMID: 37819374 Free PMC article.
-
Serving up whatever you wish: CRISPR-base editing generates novel cancer-restricted antigens for immunotherapy.Genes Immun. 2023 Dec;24(6):292-294. doi: 10.1038/s41435-023-00227-6. Epub 2023 Dec 11. Genes Immun. 2023. PMID: 38082155 No abstract available.
References
-
- Asano, T., Boisson B., Onodi F., Matuozzo D., Moncada-Velez M., Maglorius Renkilaraj M.R.L., Zhang P., Meertens L., Bolze A., Materna M., et al. . 2021. X-linked recessive TLR7 deficiency in ∼1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 6:eabl4348. 10.1126/sciimmunol.abl4348 - DOI - PMC - PubMed
-
- Baroni, M.L., Sanchez Martinez D., Gutierrez Aguera F., Roca Ho H., Castella M., Zanetti S.R., Velasco Hernandez T., Diaz de la Guardia R., Castano J., Anguita E., et al. . 2020. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J. Immunother. Cancer. 8:e000845. 10.1136/jitc-2020-000845 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
