

Targeting MCL-1 triggers DNA damage and an anti-proliferative response independent from apoptosis induction
- PMID: 37773750
- PMCID: PMC10787359
- DOI: 10.1016/j.celrep.2023.113176
Targeting MCL-1 triggers DNA damage and an anti-proliferative response independent from apoptosis induction
Abstract
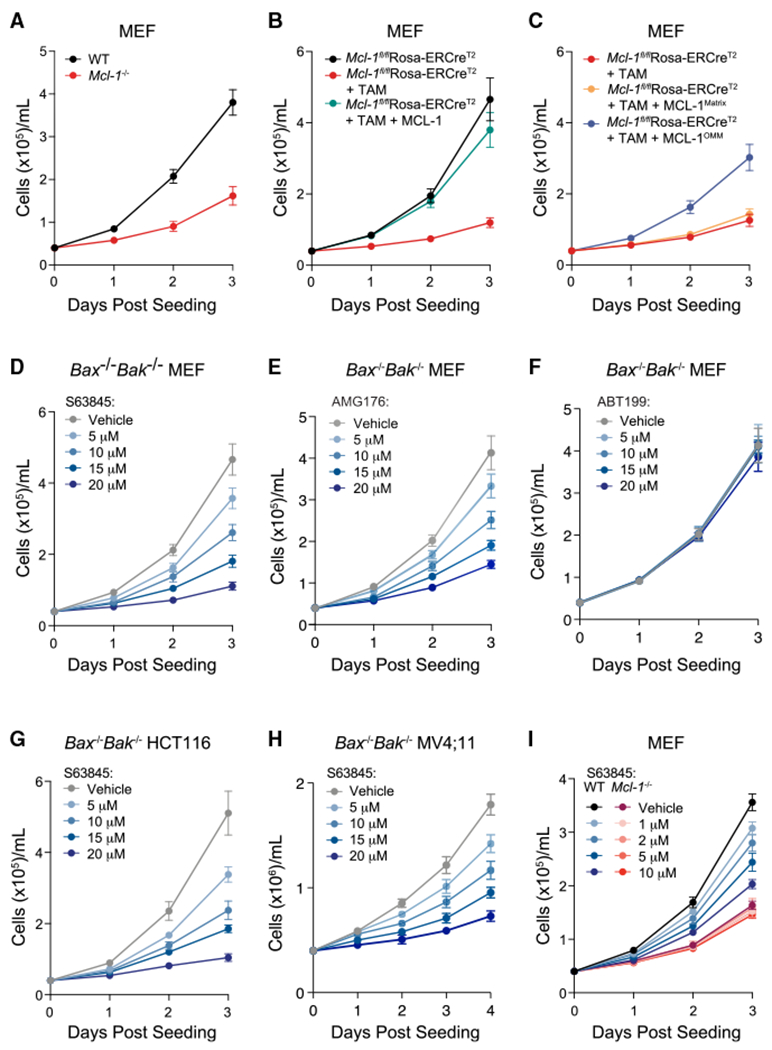
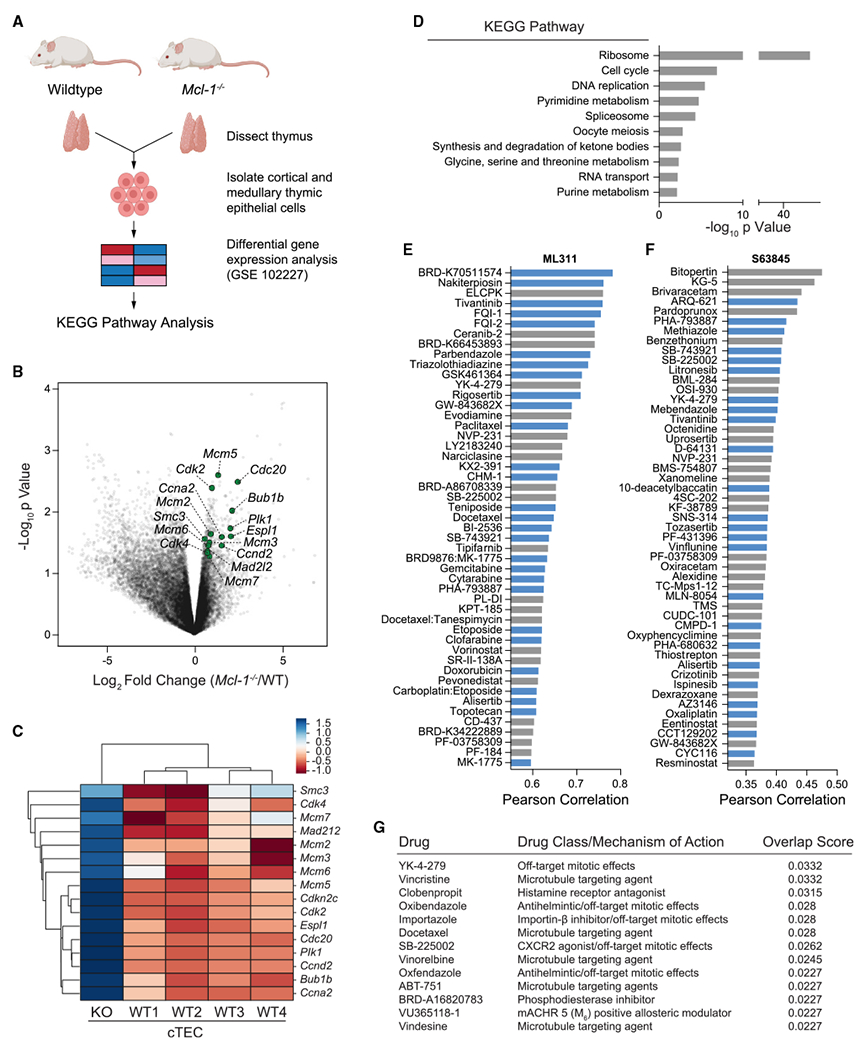
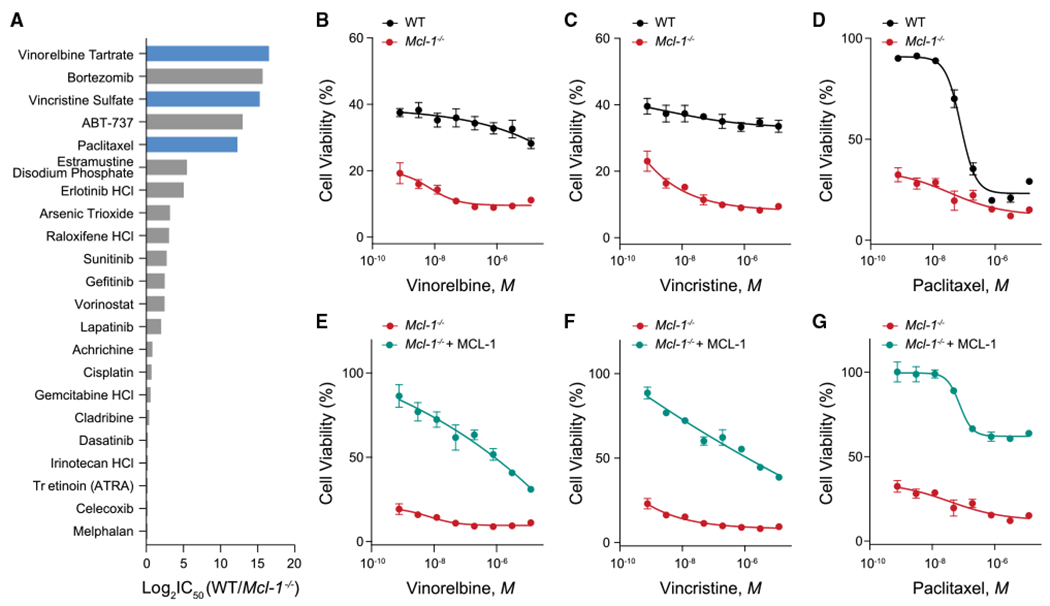
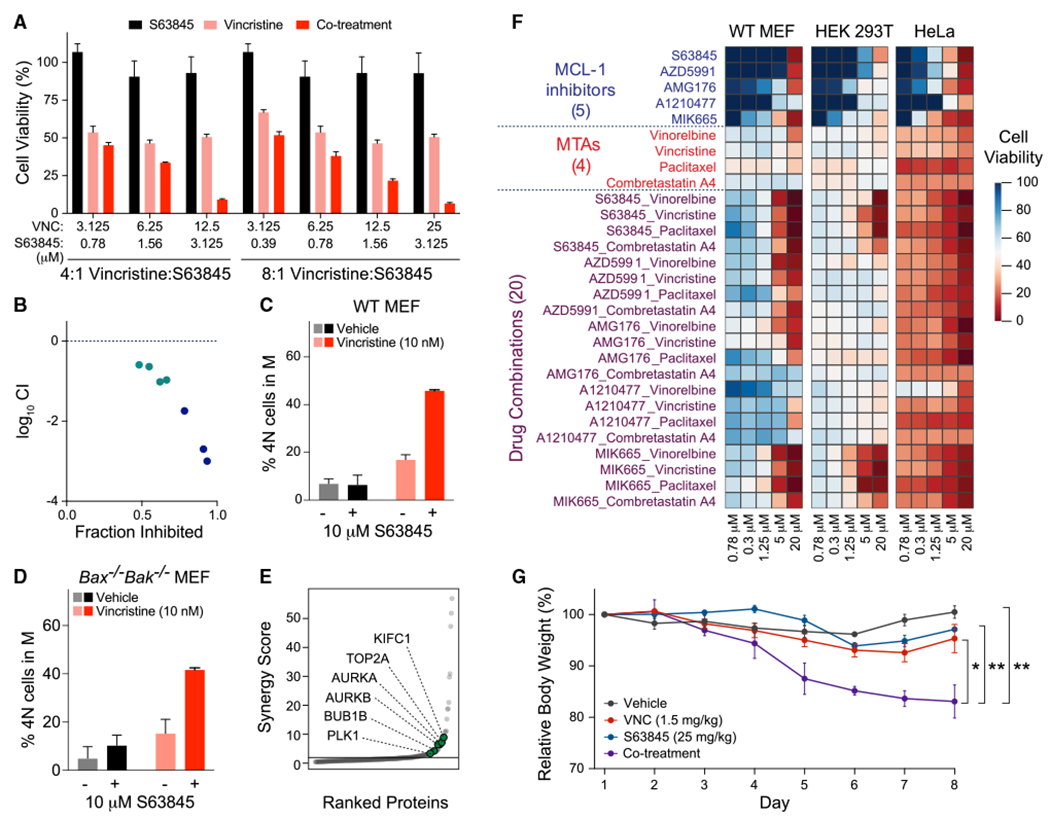
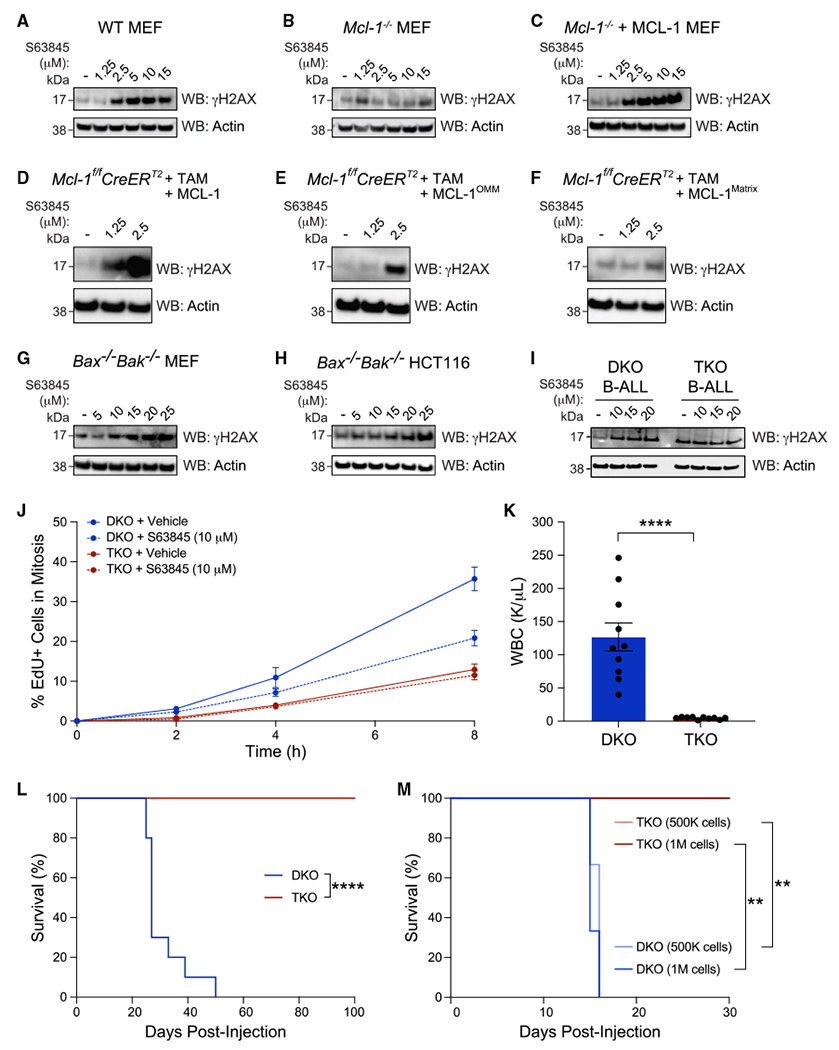
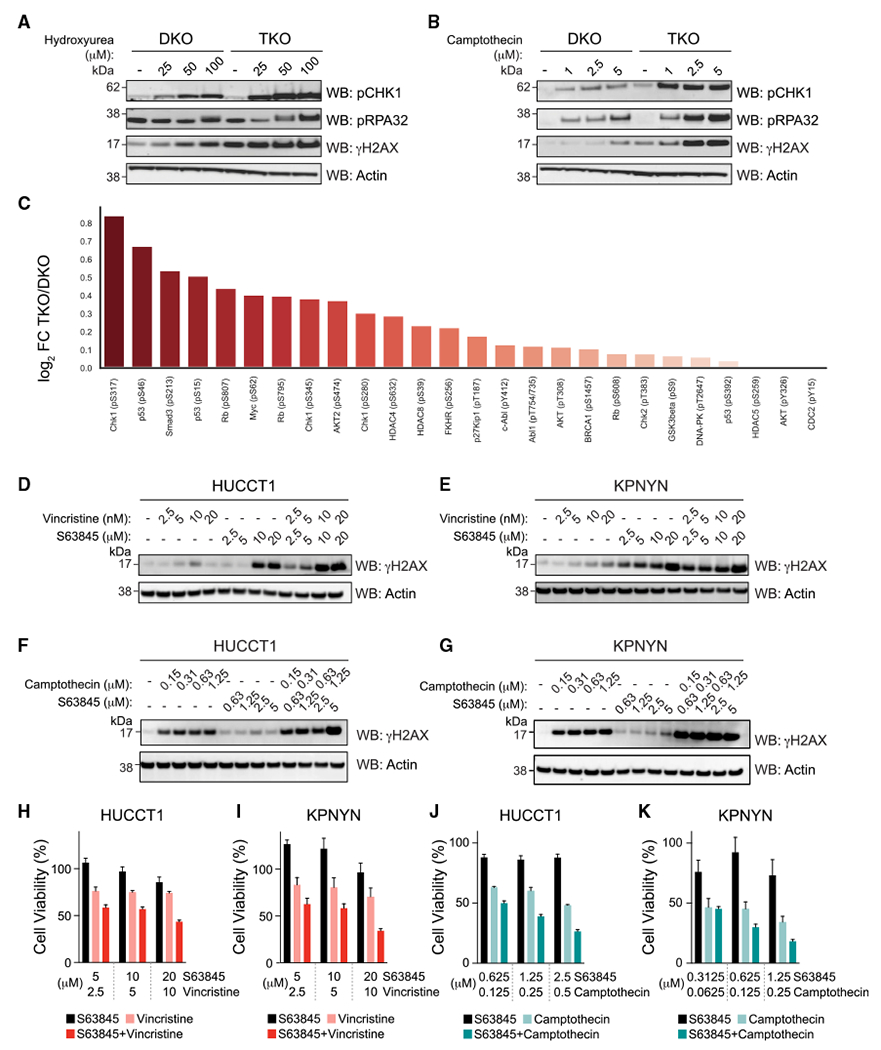
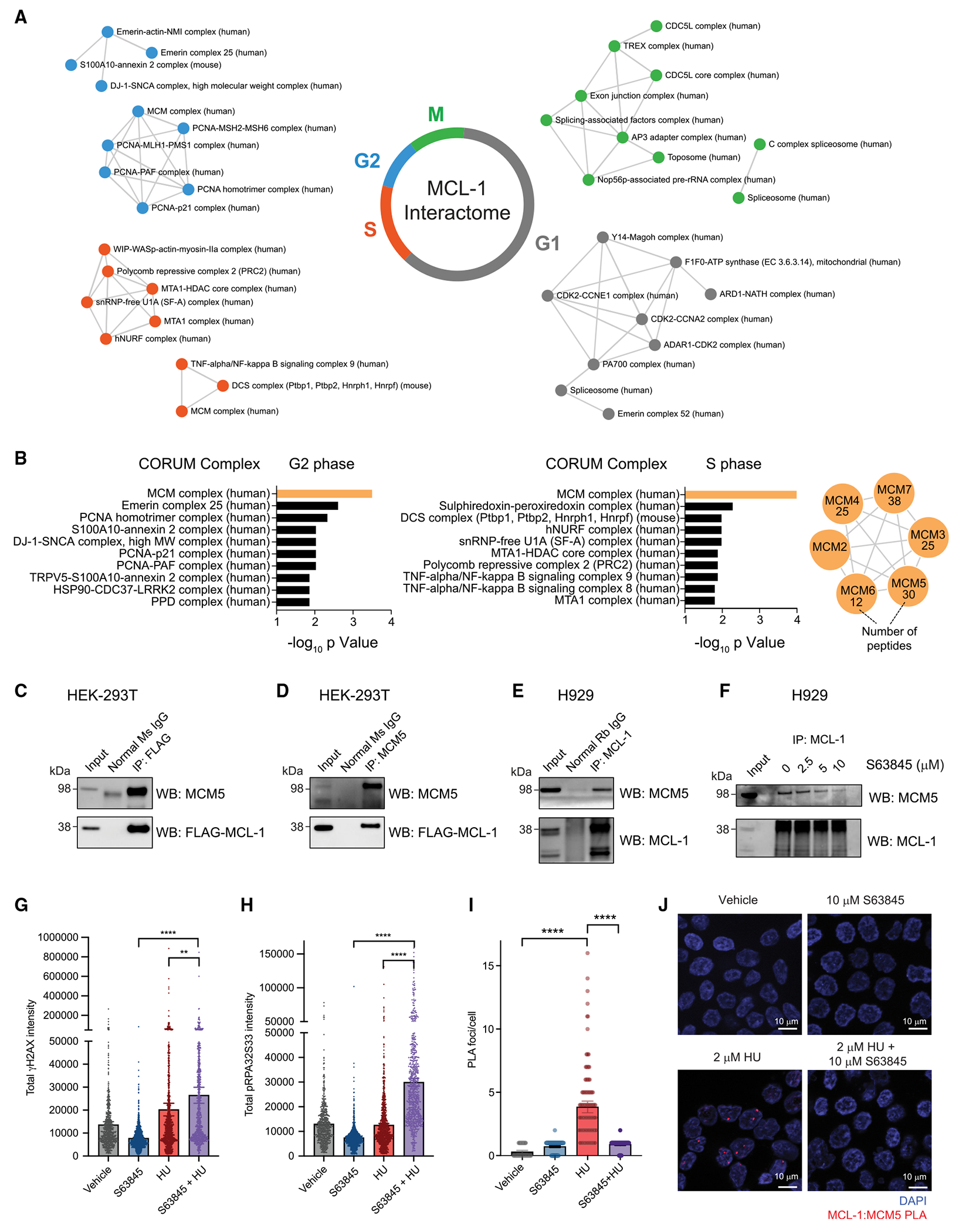
MCL-1 is a high-priority target due to its dominant role in the pathogenesis and chemoresistance of cancer, yet clinical trials of MCL-1 inhibitors are revealing toxic side effects. MCL-1 biology is complex, extending beyond apoptotic regulation and confounded by its multiple isoforms, its domains of unresolved structure and function, and challenges in distinguishing noncanonical activities from the apoptotic response. We find that, in the presence or absence of an intact mitochondrial apoptotic pathway, genetic deletion or pharmacologic targeting of MCL-1 induces DNA damage and retards cell proliferation. Indeed, the cancer cell susceptibility profile of MCL-1 inhibitors better matches that of anti-proliferative than pro-apoptotic drugs, expanding their potential therapeutic applications, including synergistic combinations, but heightening therapeutic window concerns. Proteomic profiling provides a resource for mechanistic dissection and reveals the minichromosome maintenance DNA helicase as an interacting nuclear protein complex that links MCL-1 to the regulation of DNA integrity and cell-cycle progression.
Keywords: BCL-2 family; CP: Cancer; CP: Molecular biology; DNA damage; MCL-1; apoptosis; cancer; cell cycle; cell proliferation; chemotherapy; minichrosome maintenance complex; proteomics.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Reynolds JE, Yang T, Qian L, Jenkinson JD, Zhou P, Eastman A, and Craig RW (1994). Mcl-1, a member of the Bcl-2 family, delays apoptosis induced by c-Myc overexpression in Chinese hamster ovary cells. Cancer Res. 54, 6348–6352. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
