

Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration
- PMID: 37774679
- PMCID: PMC10697236
- DOI: 10.1016/j.cell.2023.08.038
Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration
Abstract
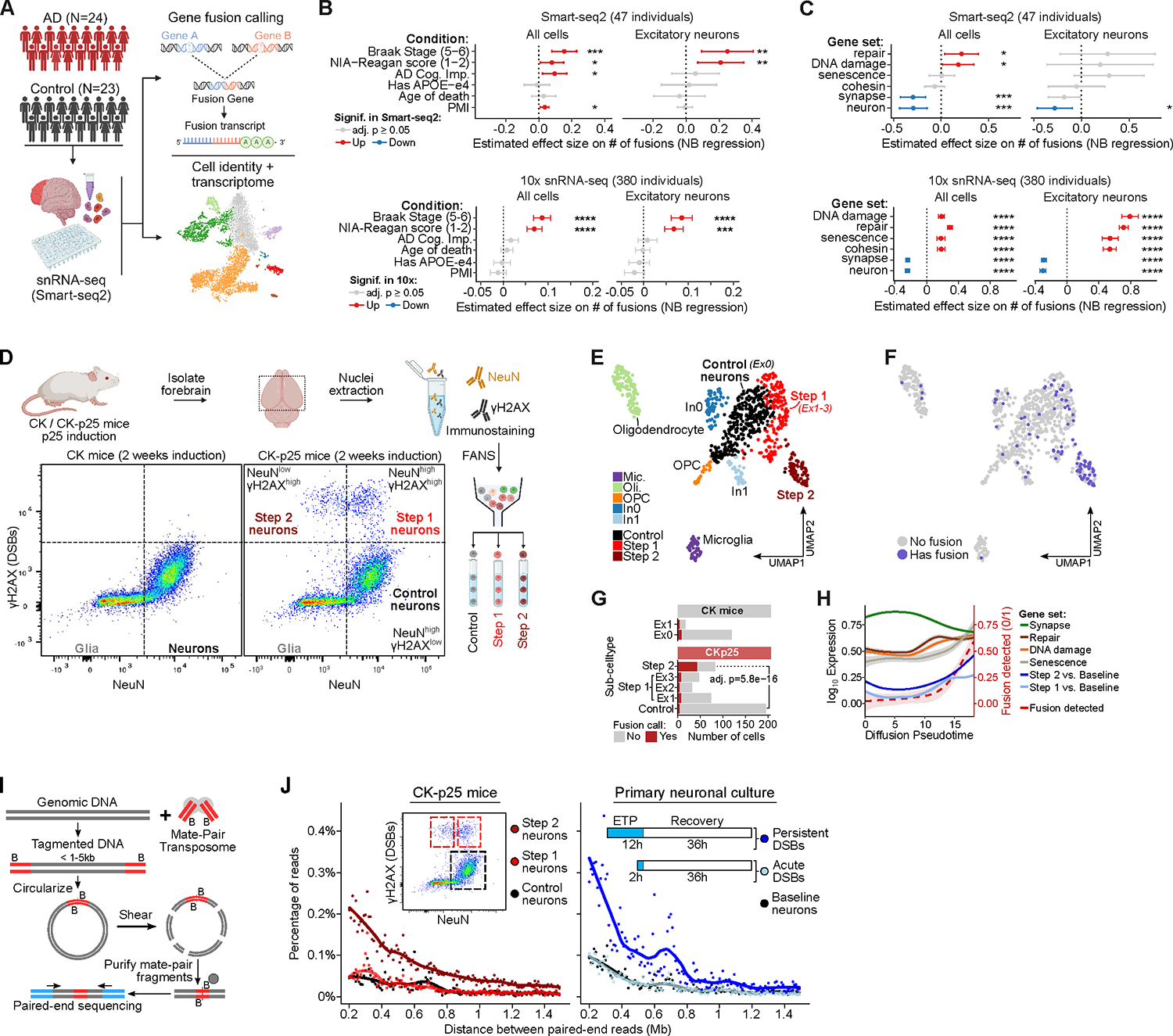
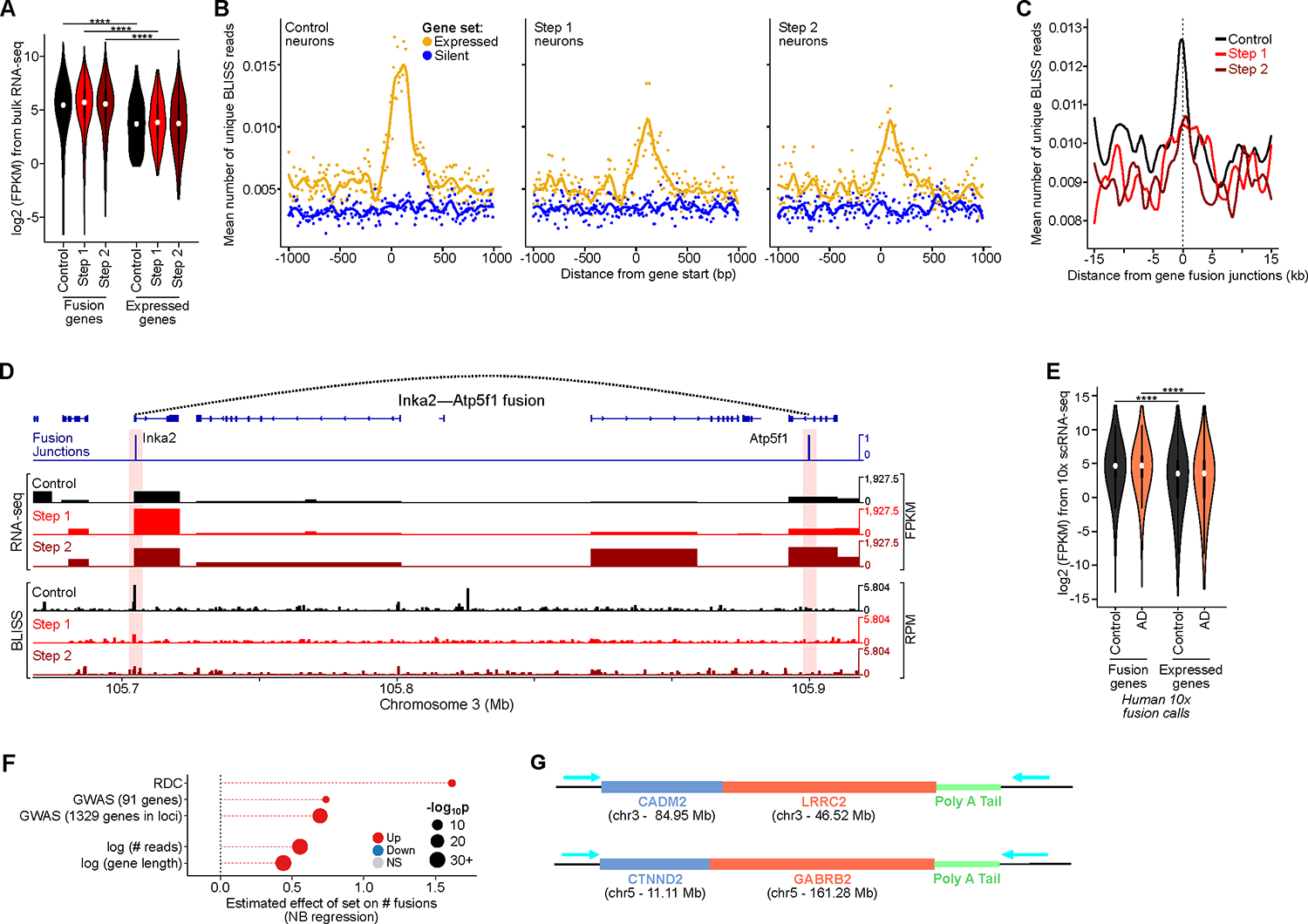
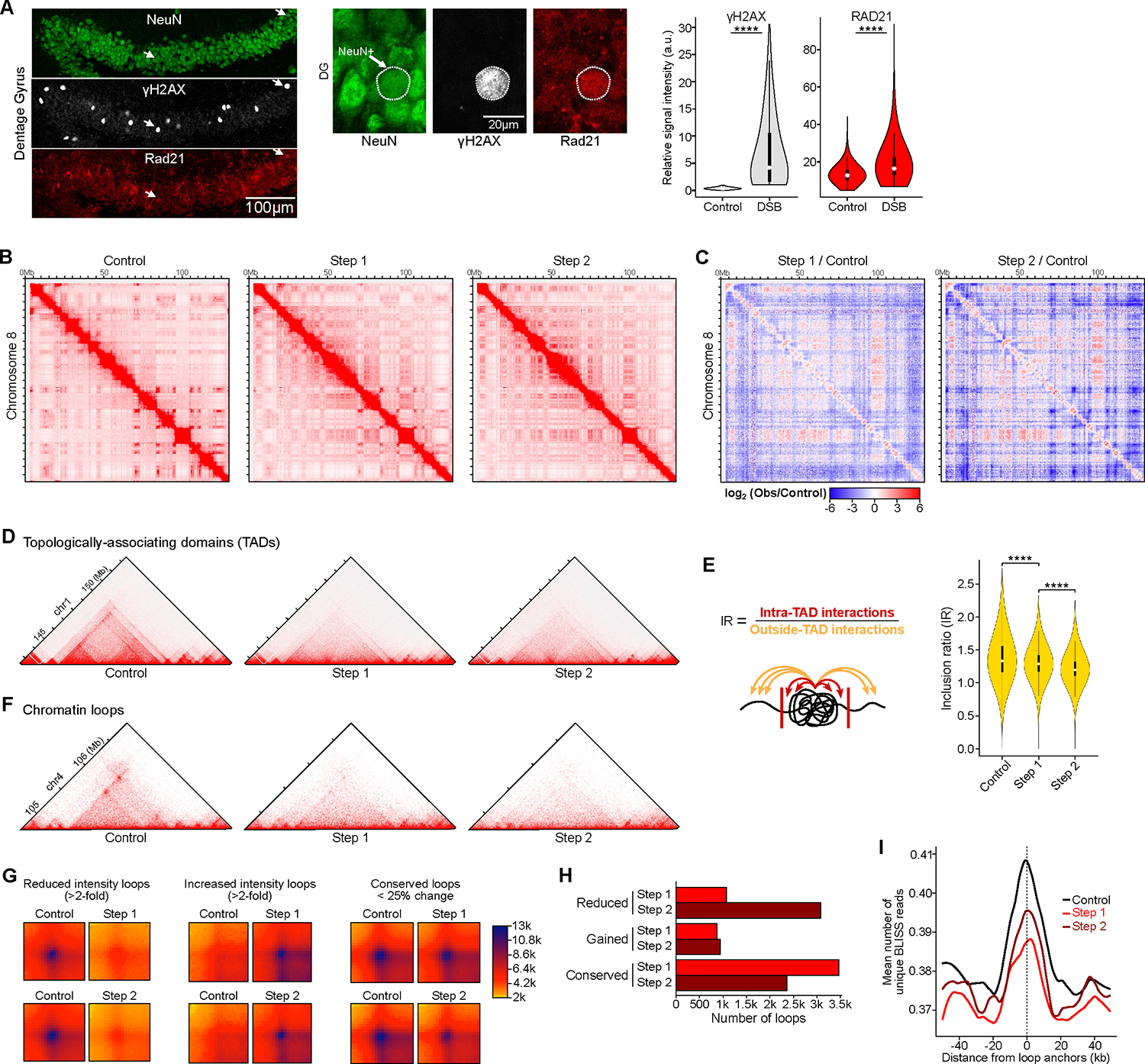
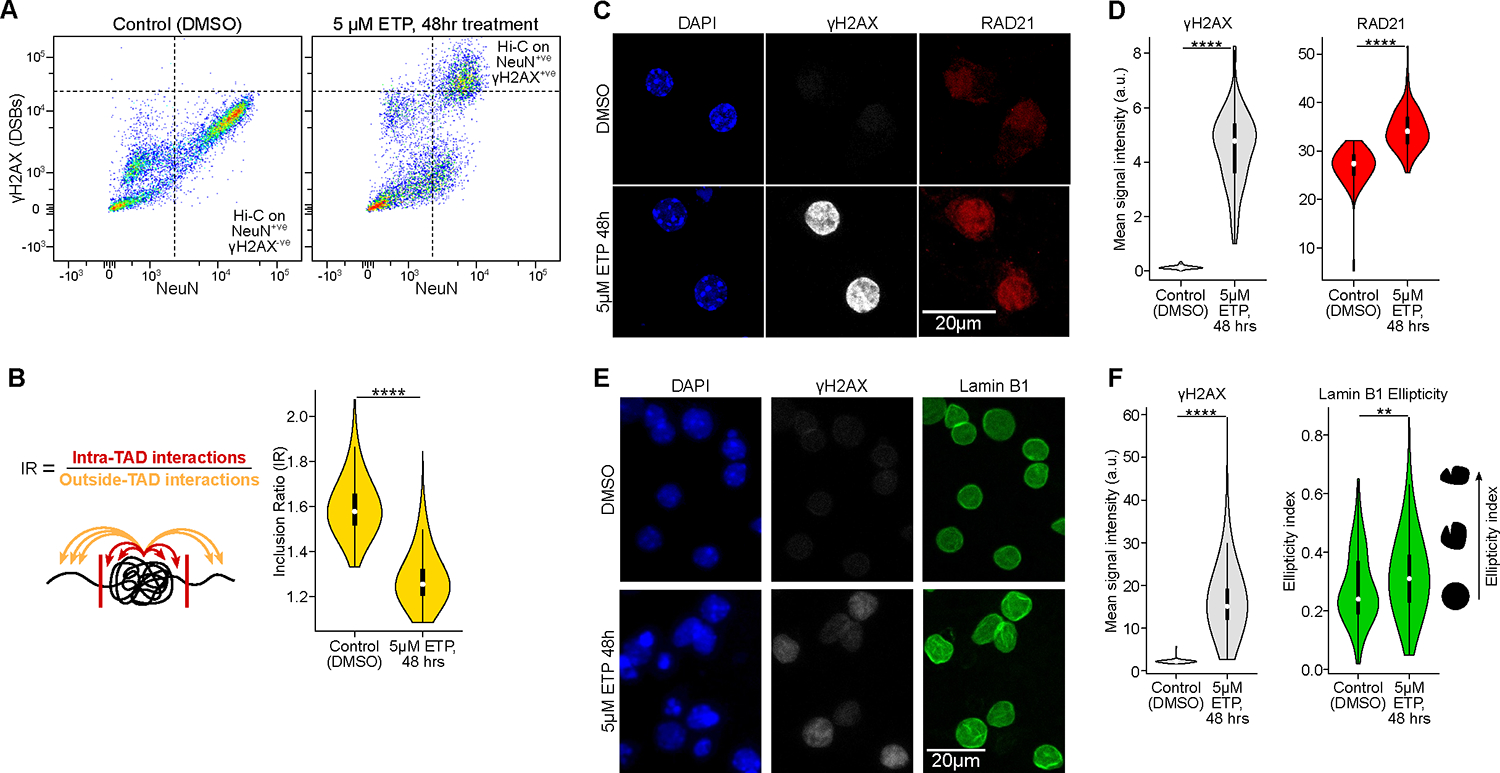
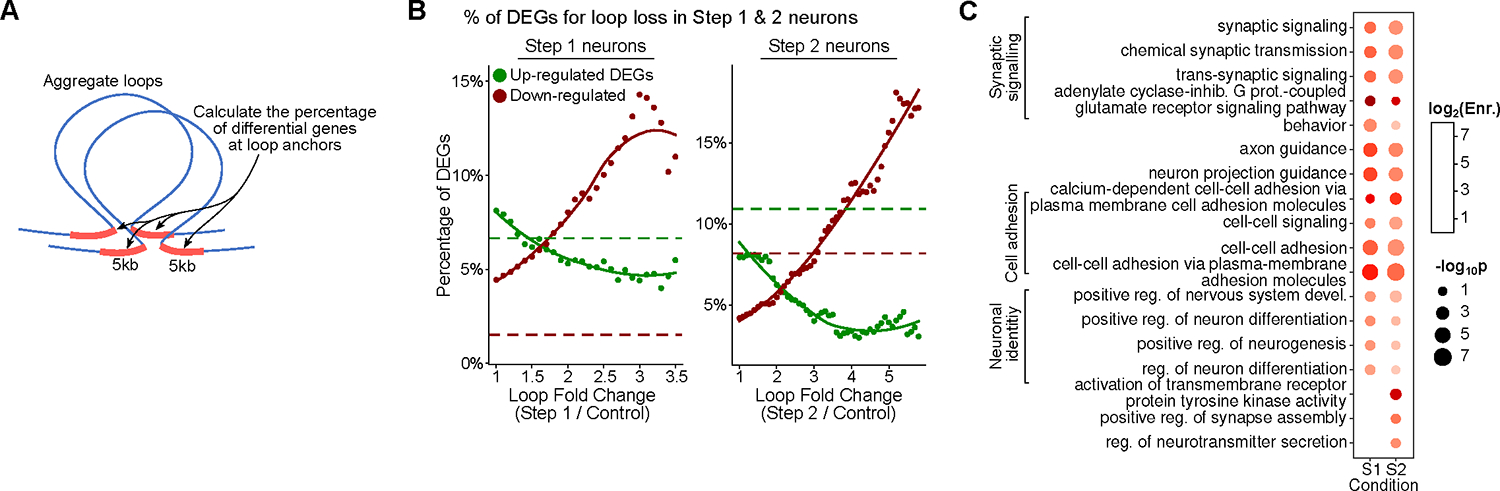
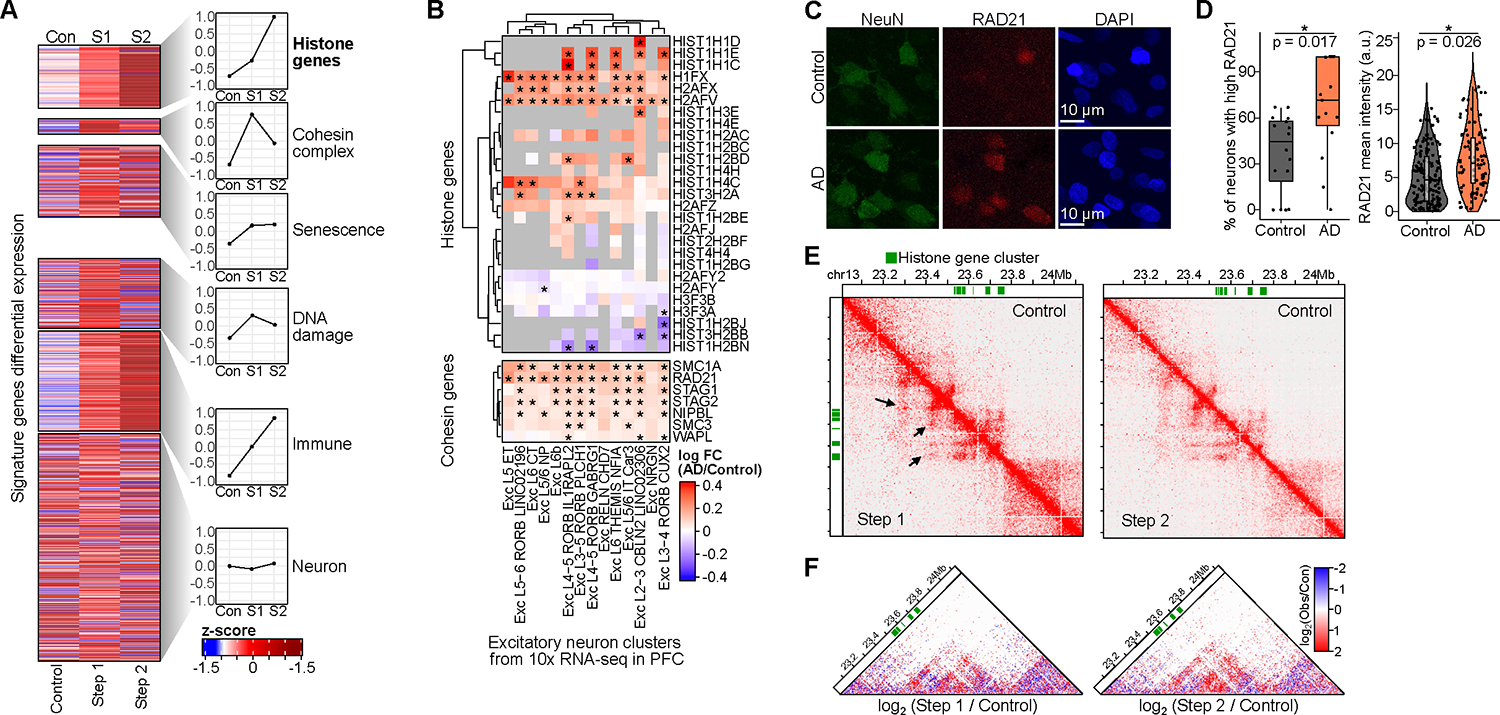
Persistent DNA double-strand breaks (DSBs) in neurons are an early pathological hallmark of neurodegenerative diseases including Alzheimer's disease (AD), with the potential to disrupt genome integrity. We used single-nucleus RNA-seq in human postmortem prefrontal cortex samples and found that excitatory neurons in AD were enriched for somatic mosaic gene fusions. Gene fusions were particularly enriched in excitatory neurons with DNA damage repair and senescence gene signatures. In addition, somatic genome structural variations and gene fusions were enriched in neurons burdened with DSBs in the CK-p25 mouse model of neurodegeneration. Neurons enriched for DSBs also had elevated levels of cohesin along with progressive multiscale disruption of the 3D genome organization aligned with transcriptional changes in synaptic, neuronal development, and histone genes. Overall, this study demonstrates the disruption of genome stability and the 3D genome organization by DSBs in neurons as pathological steps in the progression of neurodegenerative diseases.
Keywords: 3D genome organization; Alzheimer’s disease; DNA double-strand breaks; epigenome; genome rearrangements; genomic mosaicism; neurodegeneration; senescence; structural variations; transcriptome.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests L.-H.T. is a member of the Scientific Advisory Board of Cognito Therapeutics, 4M Therapeutics, Cell Signaling Technology, and Souvien Therapeutics, which has no association to the work described in this manuscript.
Figures
Comment in
-
The AD odyssey 2023: Tales of single cell.Cell. 2023 Sep 28;186(20):4257-4259. doi: 10.1016/j.cell.2023.09.001. Cell. 2023. PMID: 37774675
References
Publication types
MeSH terms
Substances
Grants and funding
- P30 AG072975/AG/NIA NIH HHS/United States
- R01 AG058002/AG/NIA NIH HHS/United States
- R01 NS051874/NS/NINDS NIH HHS/United States
- U01 AG061356/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- T32 GM087237/GM/NIGMS NIH HHS/United States
- U01 NS110453/NS/NINDS NIH HHS/United States
- R37 NS051874/NS/NINDS NIH HHS/United States
- RF1 AG054012/AG/NIA NIH HHS/United States
- U01 AG046152/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- R01 NS102730/NS/NINDS NIH HHS/United States
- K99 AG073466/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
