

Receptor-interacting protein kinase 2 (RIPK2) profoundly contributes to post-stroke neuroinflammation and behavioral deficits with microglia as unique perpetrators
- PMID: 37777791
- PMCID: PMC10543871
- DOI: 10.1186/s12974-023-02907-6
Receptor-interacting protein kinase 2 (RIPK2) profoundly contributes to post-stroke neuroinflammation and behavioral deficits with microglia as unique perpetrators
Abstract
Background: Receptor-interacting protein kinase 2 (RIPK2) is a serine/threonine kinase whose activity propagates inflammatory signaling through its association with pattern recognition receptors (PRRs) and subsequent TAK1, NF-κB, and MAPK pathway activation. After stroke, dead and dying cells release a host of damage-associated molecular patterns (DAMPs) that activate PRRs and initiate a robust inflammatory response. We hypothesize that RIPK2 plays a damaging role in the progression of stroke injury by enhancing the neuroinflammatory response to stroke and that global genetic deletion or microglia-specific conditional deletion of Ripk2 will be protective following ischemic stroke.
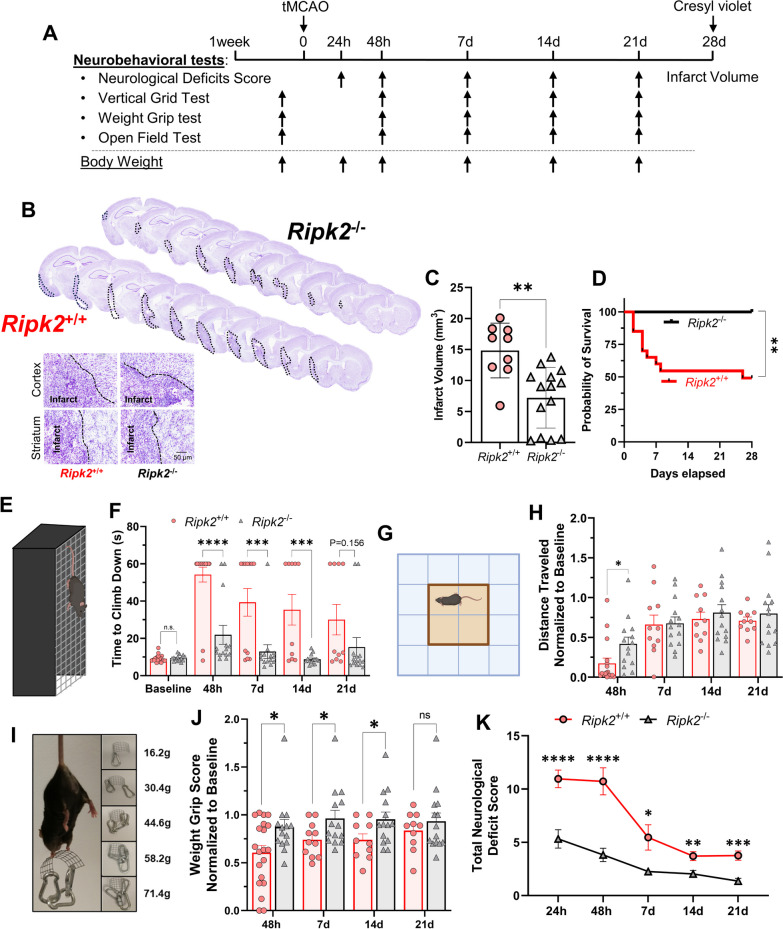
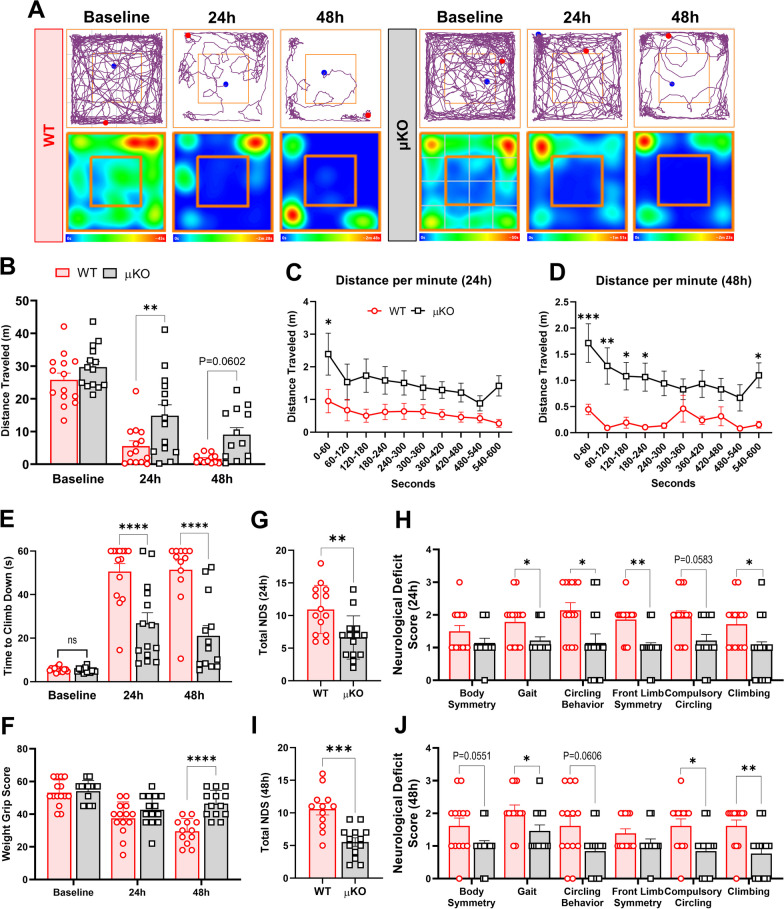
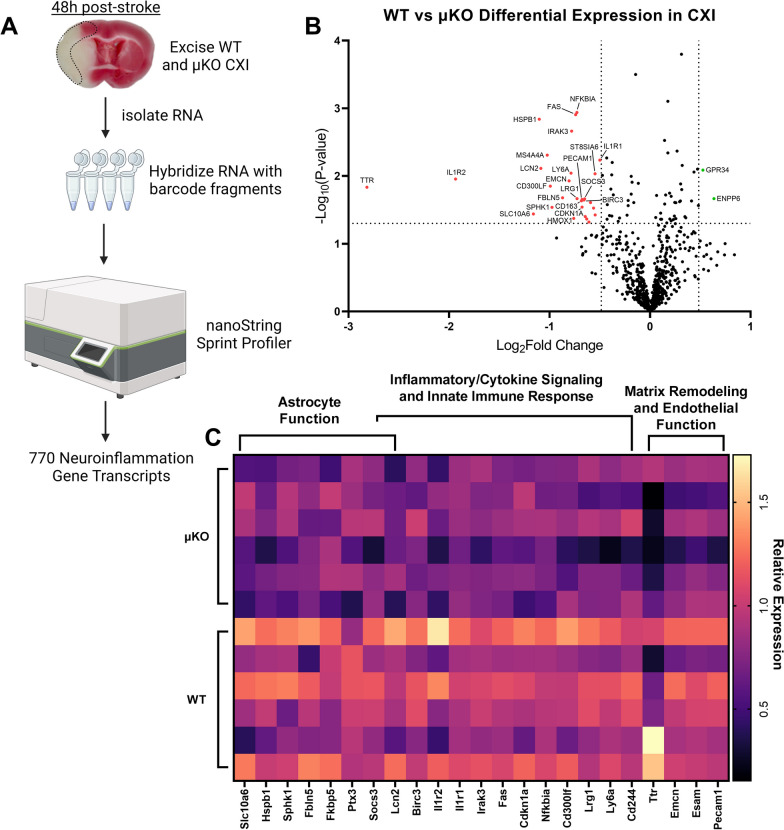
Methods: Adult (3-6 months) male mice were subjected to 45 min of transient middle cerebral artery occlusion (tMCAO) followed by 24 h, 48 h, or 28 days of reperfusion. Aged male and female mice (18-24 months) were subjected to permanent ischemic stroke and sacrificed 48 h later. Infarct volumes were calculated using TTC staining (24-48 h) or Cresyl violet staining (28d). Sensorimotor tests (weight grip, vertical grid, and open field) were performed at indicated timepoints. Blood-brain barrier (BBB) damage, tight junction proteins, matrix metalloproteinase-9 (MMP-9), and neuroinflammatory markers were assessed via immunoblotting, ELISA, immunohistochemistry, and RT-qPCR. Differential gene expression profiles were generated through bulk RNA sequencing and nanoString®.
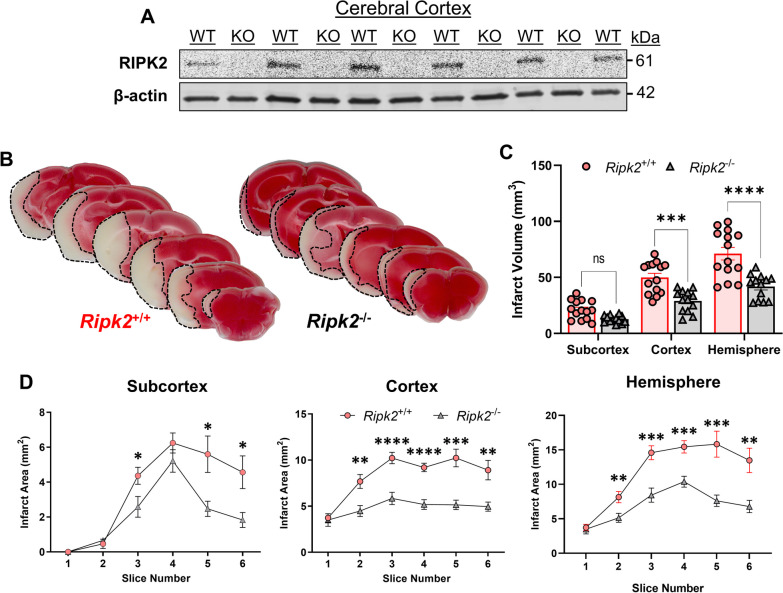
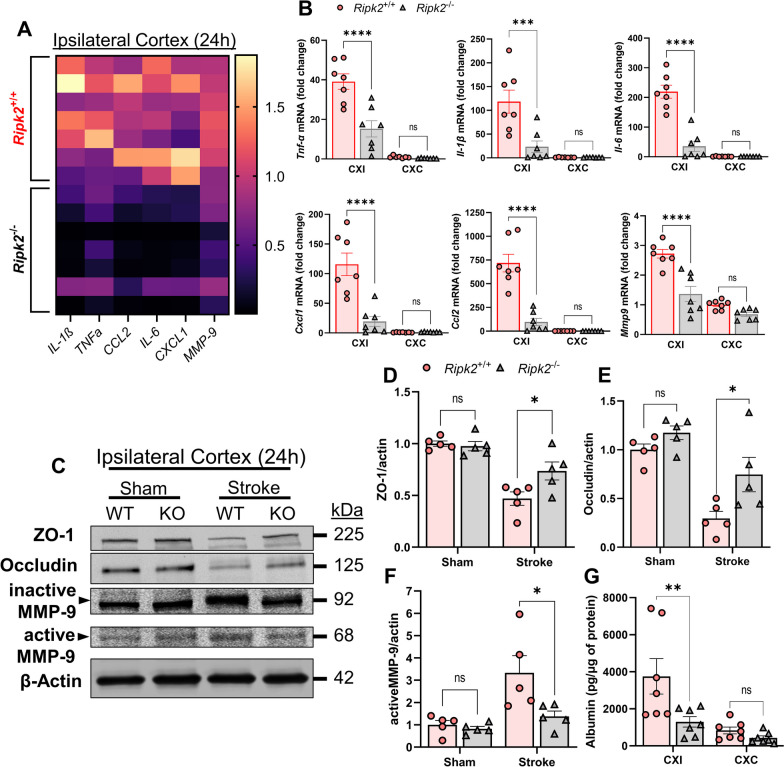
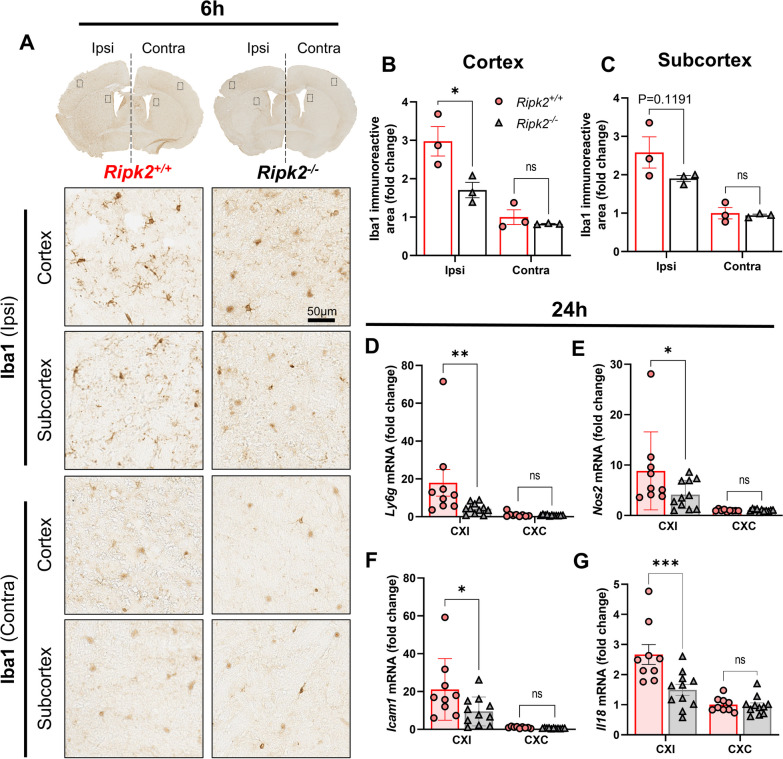
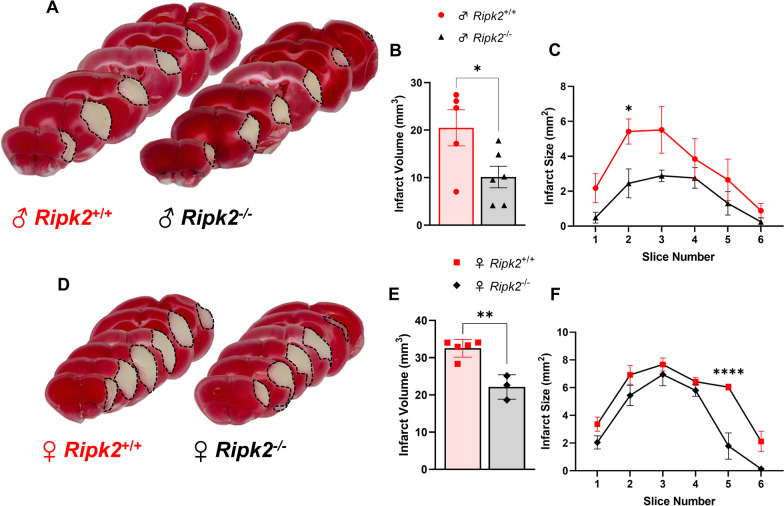
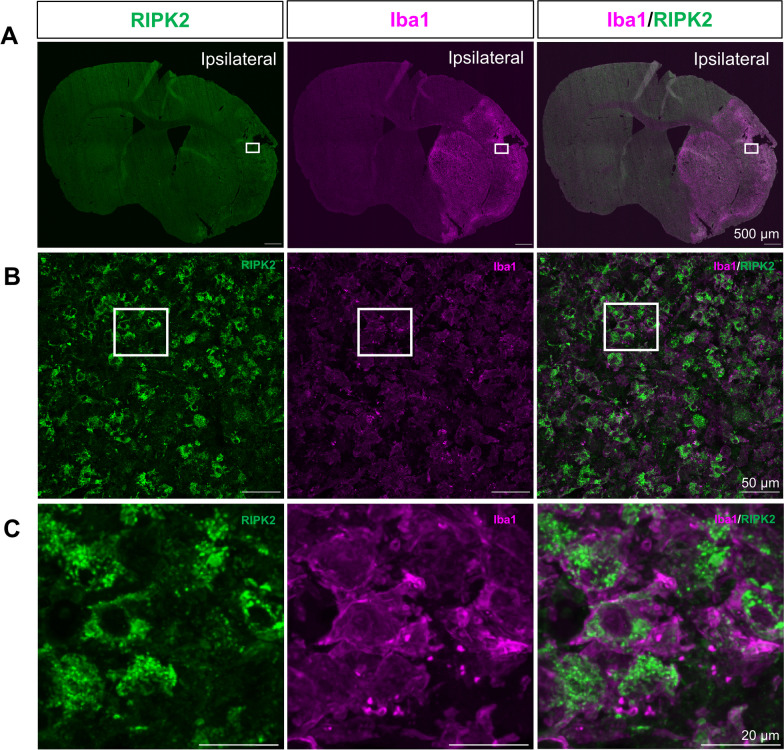
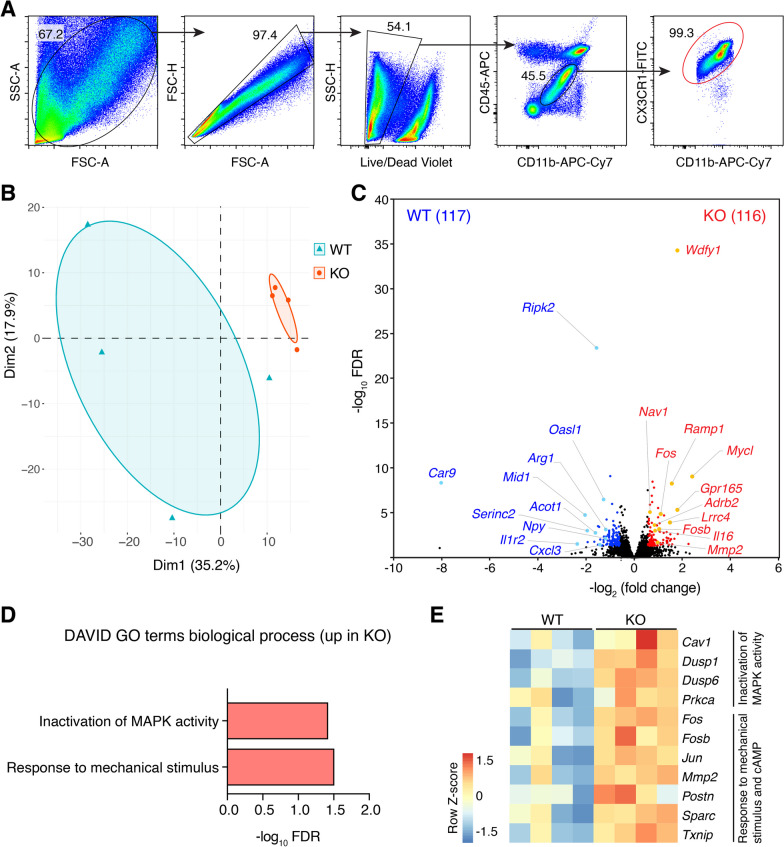
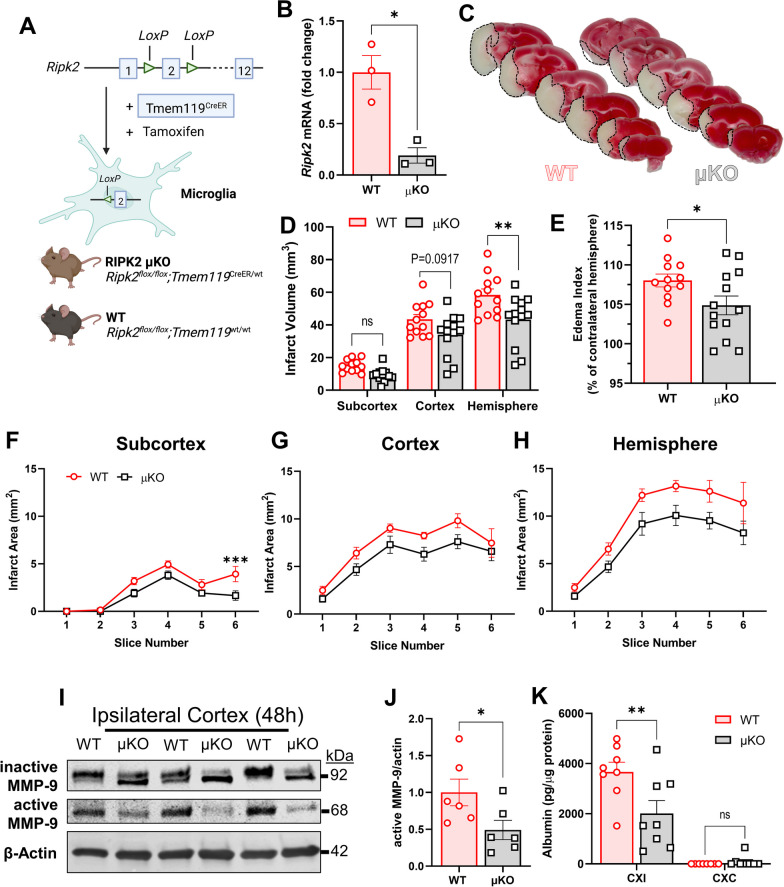
Results: Global genetic deletion of Ripk2 resulted in decreased infarct sizes and reduced neuroinflammatory markers 24 h after stroke compared to wild-type controls. Ripk2 global deletion also improved both acute and long-term behavioral outcomes with powerful effects on reducing infarct volume and mortality at 28d post-stroke. Conditional deletion of microglial Ripk2 (mKO) partially recapitulated our results in global Ripk2 deficient mice, showing reductive effects on infarct volume and improved behavioral outcomes within 48 h of injury. Finally, bulk transcriptomic profiling and nanoString data demonstrated that Ripk2 deficiency in microglia decreases genes associated with MAPK and NF-κB signaling, dampening the neuroinflammatory response after stroke injury by reducing immune cell activation and peripheral immune cell invasion.
Conclusions: These results reveal a hitherto unknown role for RIPK2 in the pathogenesis of ischemic stroke injury, with microglia playing a distinct role. This study identifies RIPK2 as a potent propagator of neuroinflammatory signaling, highlighting its potential as a therapeutic target for post-stroke intervention.
Keywords: Blood–brain barrier injury; Ischemic stroke; Microglia; Neuroinflammation; RIPK2.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Apelin-13 enhances neurofunctional recovery and suppresses neuroinflammation via the SIRT1/NF-κB axis in ischemic stroke.Cell Immunol. 2025 Jul;413:104958. doi: 10.1016/j.cellimm.2025.104958. Epub 2025 Apr 22. Cell Immunol. 2025. PMID: 40378509
-
Therapeutic Benefits of Adropin in Aged Mice After Transient Ischemic Stroke via Reduction of Blood-Brain Barrier Damage.Stroke. 2023 Jan;54(1):234-244. doi: 10.1161/STROKEAHA.122.039628. Epub 2022 Oct 28. Stroke. 2023. PMID: 36305313 Free PMC article.
-
Effects of global Ripk2 genetic deficiency in aged mice following experimental ischemic stroke.Aging Brain. 2025 Mar 29;7:100135. doi: 10.1016/j.nbas.2025.100135. eCollection 2025. Aging Brain. 2025. PMID: 40225421 Free PMC article.
-
Loureirin B protects against cerebral ischemia/reperfusion injury through modulating M1/M2 microglial polarization via STAT6 / NF-kappaB signaling pathway.Eur J Pharmacol. 2023 Aug 15;953:175860. doi: 10.1016/j.ejphar.2023.175860. Epub 2023 Jun 16. Eur J Pharmacol. 2023. PMID: 37331681 Review.
-
Ripks and Neuroinflammation.Mol Neurobiol. 2024 Sep;61(9):6771-6787. doi: 10.1007/s12035-024-03981-4. Epub 2024 Feb 13. Mol Neurobiol. 2024. PMID: 38349514 Review.
Cited by
-
Intersecting Pathways: The Role of Metabolic Dysregulation, Gastrointestinal Microbiome, and Inflammation in Acute Ischemic Stroke Pathogenesis and Outcomes.J Clin Med. 2024 Jul 21;13(14):4258. doi: 10.3390/jcm13144258. J Clin Med. 2024. PMID: 39064298 Free PMC article. Review.
-
RIPK2 Is Crucial for the Microglial Inflammatory Response to Bacterial Muramyl Dipeptide but Not to Lipopolysaccharide.Int J Mol Sci. 2024 Nov 1;25(21):11754. doi: 10.3390/ijms252111754. Int J Mol Sci. 2024. PMID: 39519307 Free PMC article.
-
Current advances on RIPK2 and its inhibitors in pathological processes: a comprehensive review.Front Mol Neurosci. 2025 May 8;18:1492807. doi: 10.3389/fnmol.2025.1492807. eCollection 2025. Front Mol Neurosci. 2025. PMID: 40406369 Free PMC article. Review.
-
Plasma biomarkers in patients with familial cavernous malformation and their first-degree relatives: a cross-sectional study.Sci Rep. 2025 Apr 2;15(1):11284. doi: 10.1038/s41598-025-91141-6. Sci Rep. 2025. PMID: 40175401 Free PMC article.
-
CMD-OPT model enables the discovery of a potent and selective RIPK2 inhibitor as preclinical candidate for the treatment of acute liver injury.Acta Pharm Sin B. 2025 Jul;15(7):3708-3724. doi: 10.1016/j.apsb.2025.05.003. Epub 2025 May 13. Acta Pharm Sin B. 2025. PMID: 40698146 Free PMC article.
References
-
- Candelario-Jalil E. Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs. 2009;10:644–654. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
