

Characterizing citrullination by mass spectrometry-based proteomics
- PMID: 37778389
- PMCID: PMC10542455
- DOI: 10.1098/rstb.2022.0237
Characterizing citrullination by mass spectrometry-based proteomics
Abstract
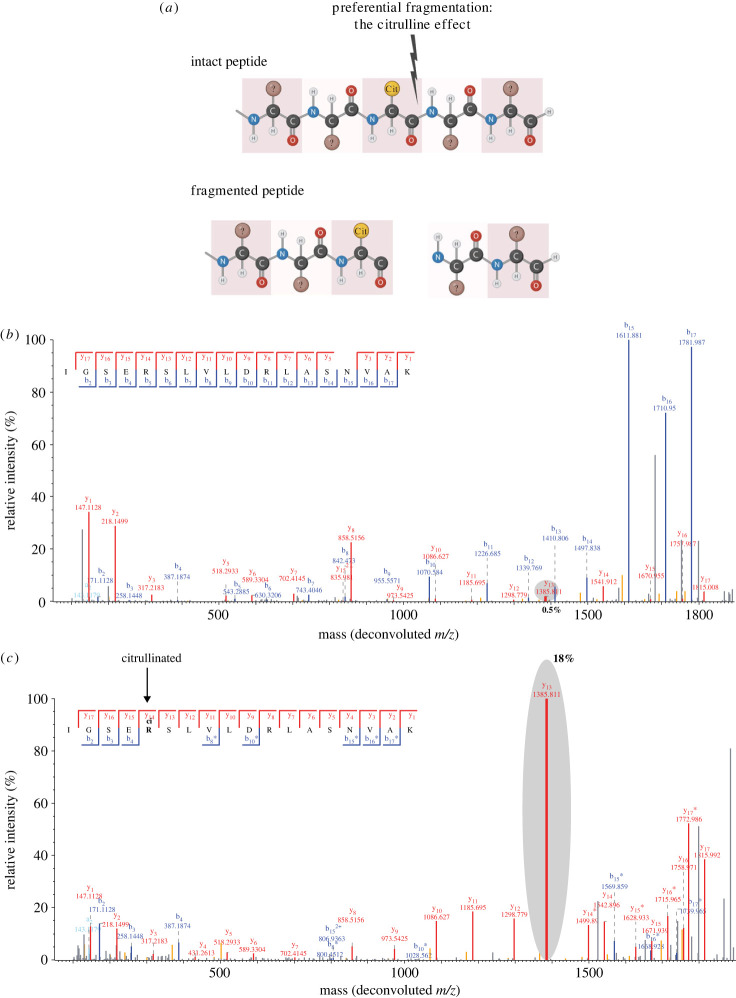
Citrullination is an important post-translational modification (PTM) of arginine, known to play a role in autoimmune disorders, innate immunity response and maintenance of stem cell potency. However, citrullination remains poorly characterized and not as comprehensively understood compared to other PTMs, such as phosphorylation and ubiquitylation. High-resolution mass spectrometry (MS)-based proteomics offers a valuable approach for studying citrullination in an unbiased manner, allowing confident identification of citrullination modification sites and distinction from deamidation events on asparagine and glutamine. MS efforts have already provided valuable insights into peptidyl arginine deaminase targeting along with site-specific information of citrullination in for example synovial fluids derived from rheumatoid arthritis patients. Still, there is unrealized potential for the wider citrullination field by applying MS-based mass spectrometry approaches for proteome-wide investigations. Here we will outline contemporary methods and current challenges for studying citrullination by MS, and discuss how the development of neoteric citrullination-specific proteomics approaches still may improve our understanding of citrullination networks. This article is part of the Theo Murphy meeting issue 'The virtues and vices of protein citrullination'.
Keywords: citrullination; mass spectrometry; post-translational modifications; proteomics.
Conflict of interest statement
We have no competing interests.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous