

Oncogenic KRAS mutation confers chemoresistance by upregulating SIRT1 in non-small cell lung cancer
- PMID: 37779142
- PMCID: PMC10618295
- DOI: 10.1038/s12276-023-01091-0
Oncogenic KRAS mutation confers chemoresistance by upregulating SIRT1 in non-small cell lung cancer
Abstract
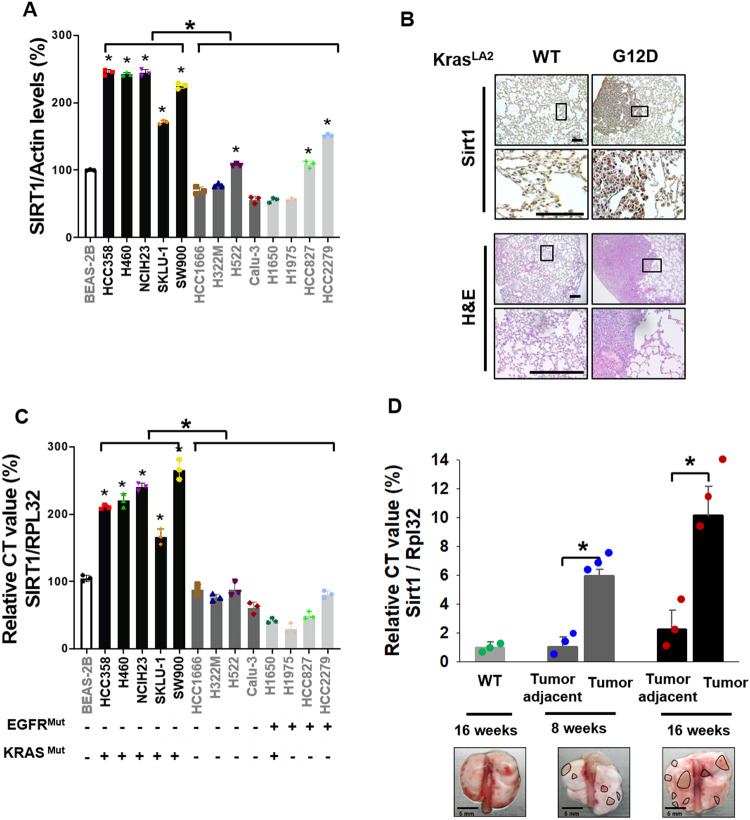
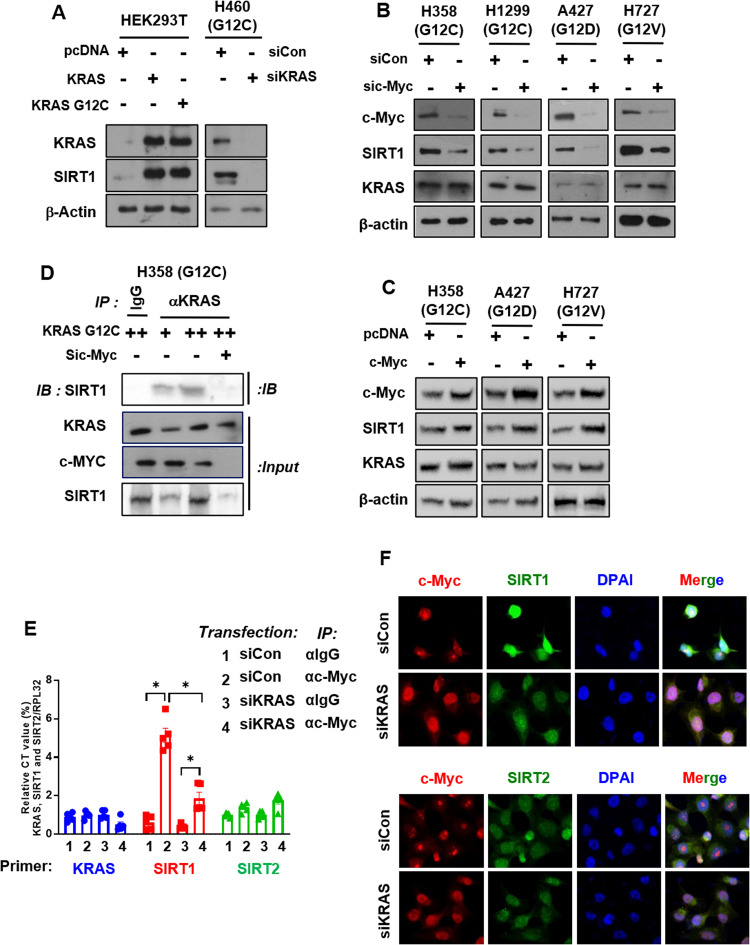
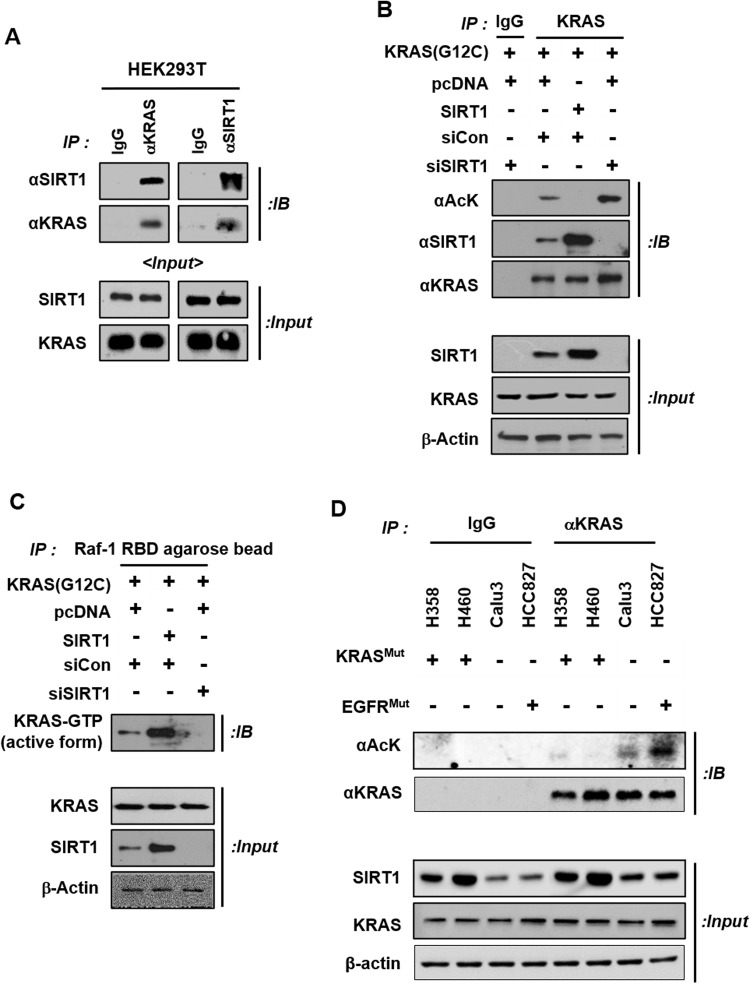
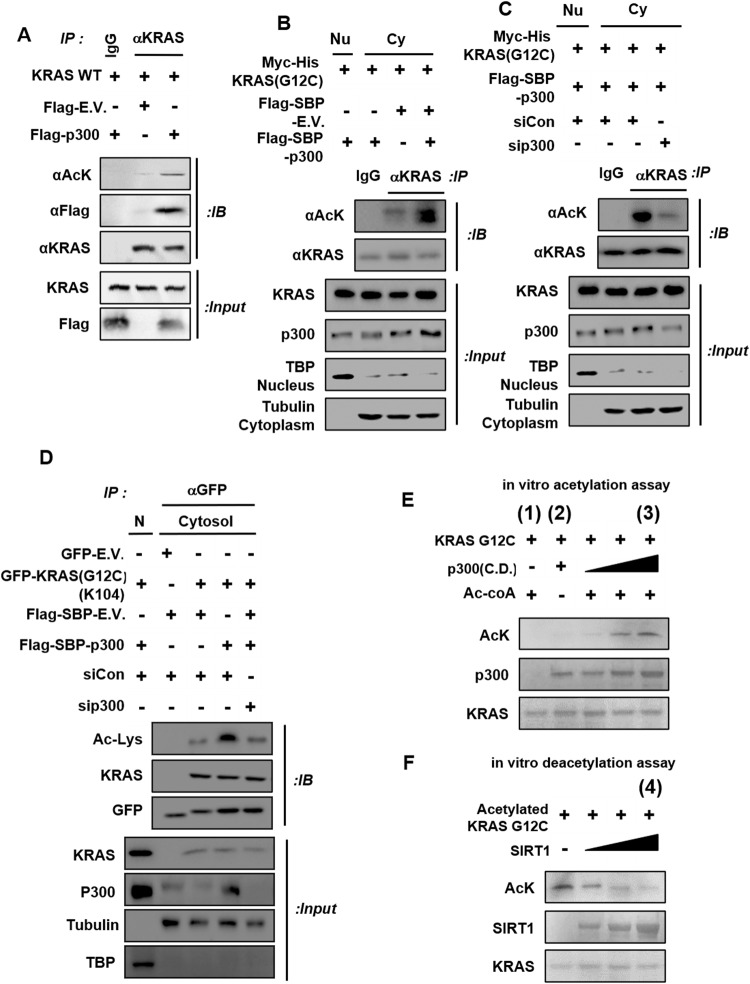
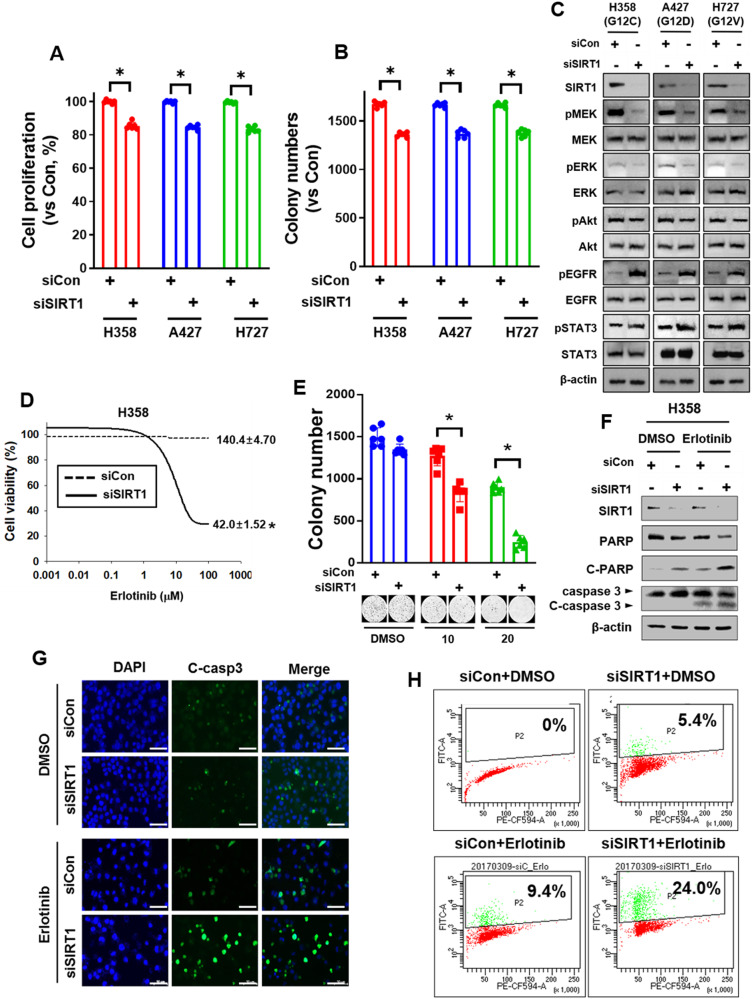
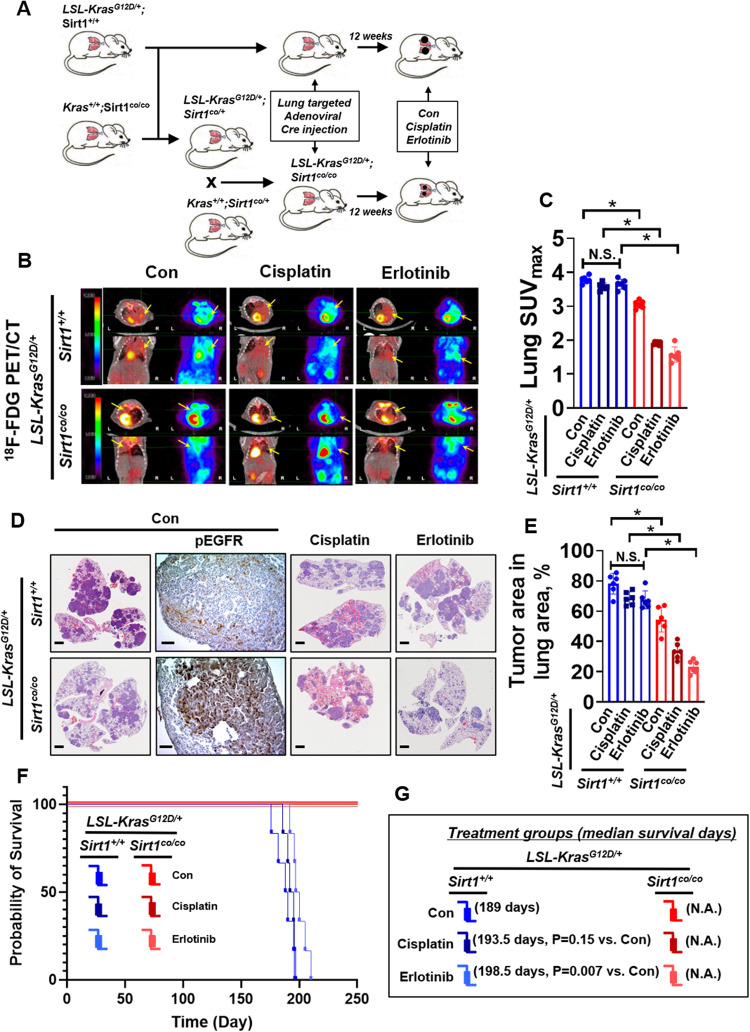
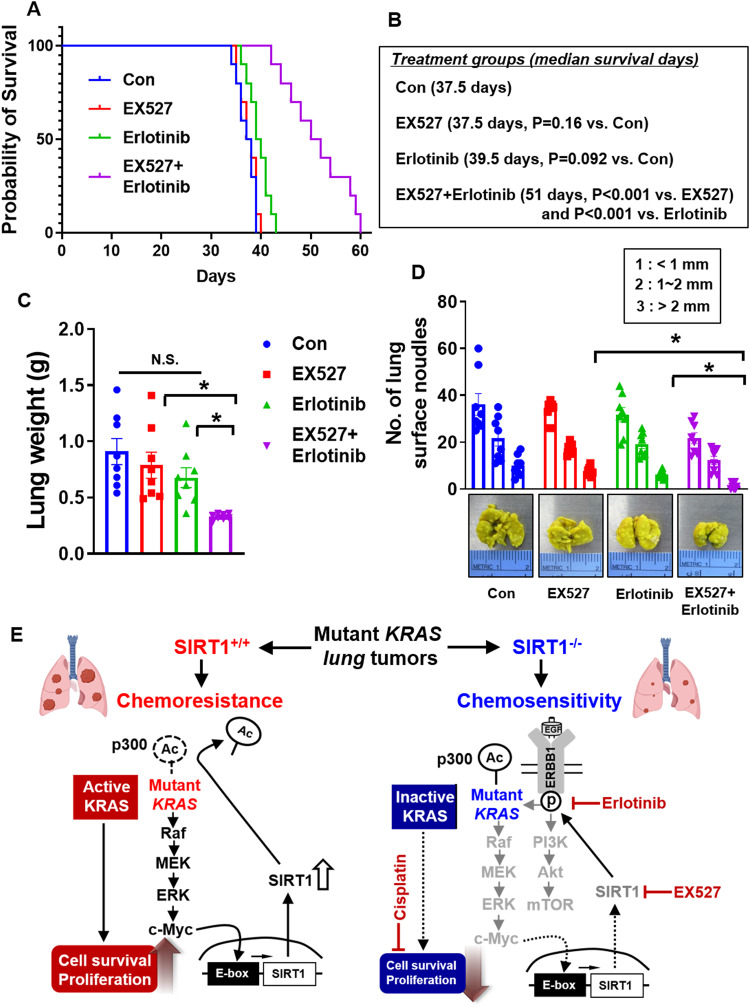
Kirsten rat sarcoma viral oncogene homologue (KRAS) is a frequent oncogenic driver of solid tumors, including non-small cell lung cancer (NSCLC). The treatment and outcomes of KRAS-mutant cancers have not been dramatically revolutionized by direct KRAS-targeted therapies because of the lack of deep binding pockets for specific small molecule inhibitors. Here, we demonstrated that the mRNA and protein levels of the class III histone deacetylase SIRT1 were upregulated by the KRASMut-Raf-MEK-c-Myc axis in KRASMut lung cancer cells and in lung tumors of a mouse model with spontaneous KrasG12D expression. KRASMut-induced SIRT1 bound to KRASMut and stably deacetylated KRASMut at lysine 104, which increased KRASMut activity. SIRT1 knockdown (K/D) or the SIRT1H363Y mutation increased KRASMut acetylation, which decreased KRASMut activity and sensitized tumors to the anticancer effects of cisplatin and erlotinib. Furthermore, in KrasG12D/+;Sirt1co/co mice, treatment with cisplatin and erlotinib robustly reduced the tumor burden and increased survival rates compared with those in spontaneous LSL-KrasG12D/+;Sirt1+/+ mice and mice in each single-drug treatment group. Then, we identified p300 as a KRASMut acetyltransferase that reinforced KRASMut lysine 104 acetylation and robustly decreased KRASMut activity. KRASMut lysine 104 acetylation by p300 and deacetylation by SIRT1 were confirmed by LC‒MS/MS. Consistent with this finding, the SIRT1 inhibitor EX527 suppressed KRASMut activity, which synergistically abolished cell proliferation and colony formation, as well as the tumor burden in KRASMut mice, when combined with cisplatin or erlotinib. Our data reveal a novel pathway critical for the regulation of KRASMut lung cancer progression and provide important evidence for the potential application of SIRT1 inhibitors and p300 activators for the combination treatment of KRASMut lung cancer patients.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
